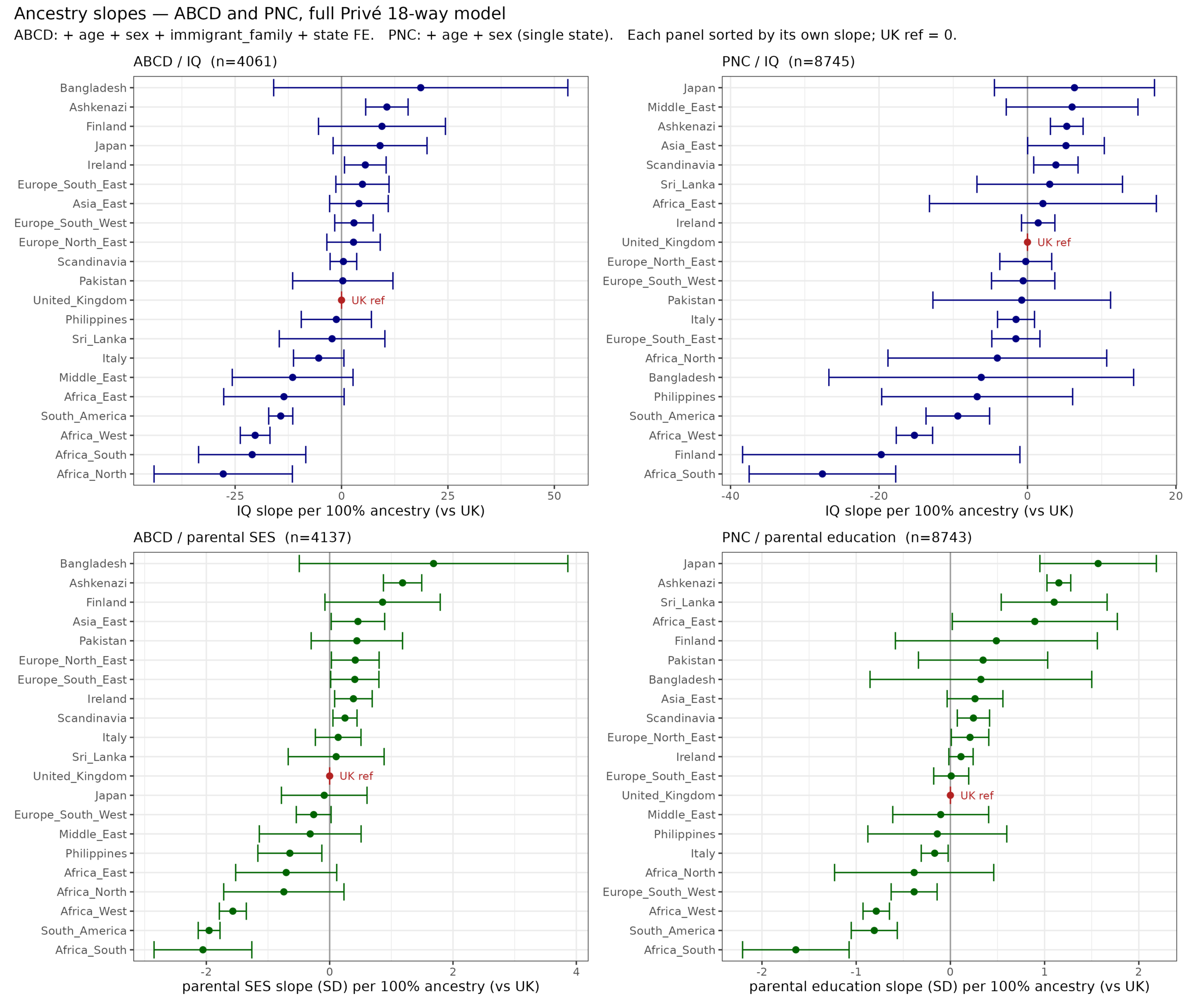
Genetic evidence of Ashkenazi intelligence
There are long standing questions about the source of Jewish achievement. Most of this achievement relates to the Ashkenazi subgroup, less so the other Jews. This tells us that the…
There are long standing questions about the source of Jewish achievement. Most of this achievement relates to the Ashkenazi subgroup, less so the other Jews. This tells us that the…

Some time ago, we published studies showing that the negative correlation between intelligence and fertility varies by other factors. Specifically, we found that it varies by religiousness and conservatism, in…
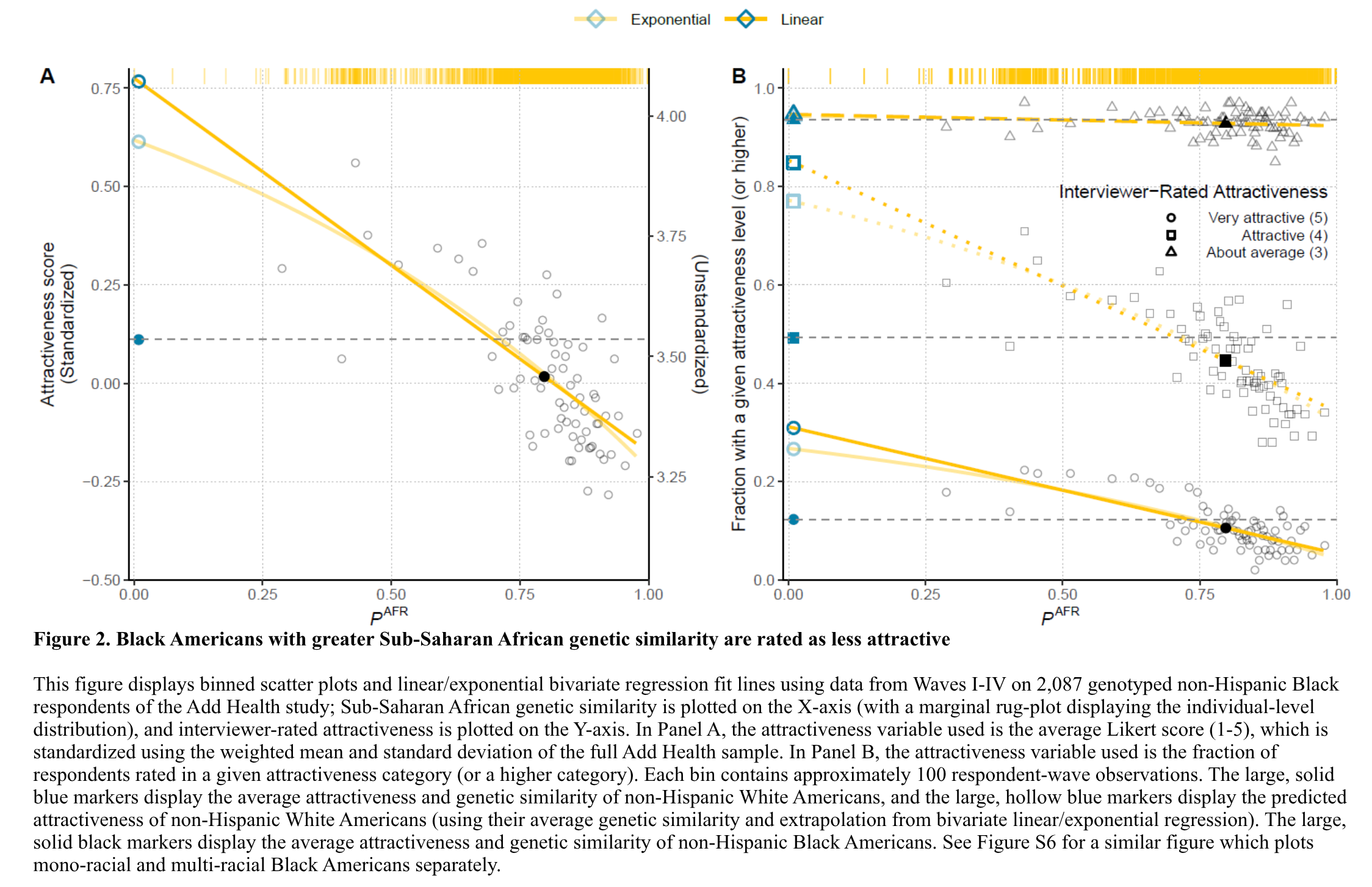
A friend sent me this study yesterday, so let's cover it here: Taddess, B., Zhang, L., & Trejo, S. (2025). Leveraging Genomic Data to Document Within-Race Attractiveness Penalties Among Black…
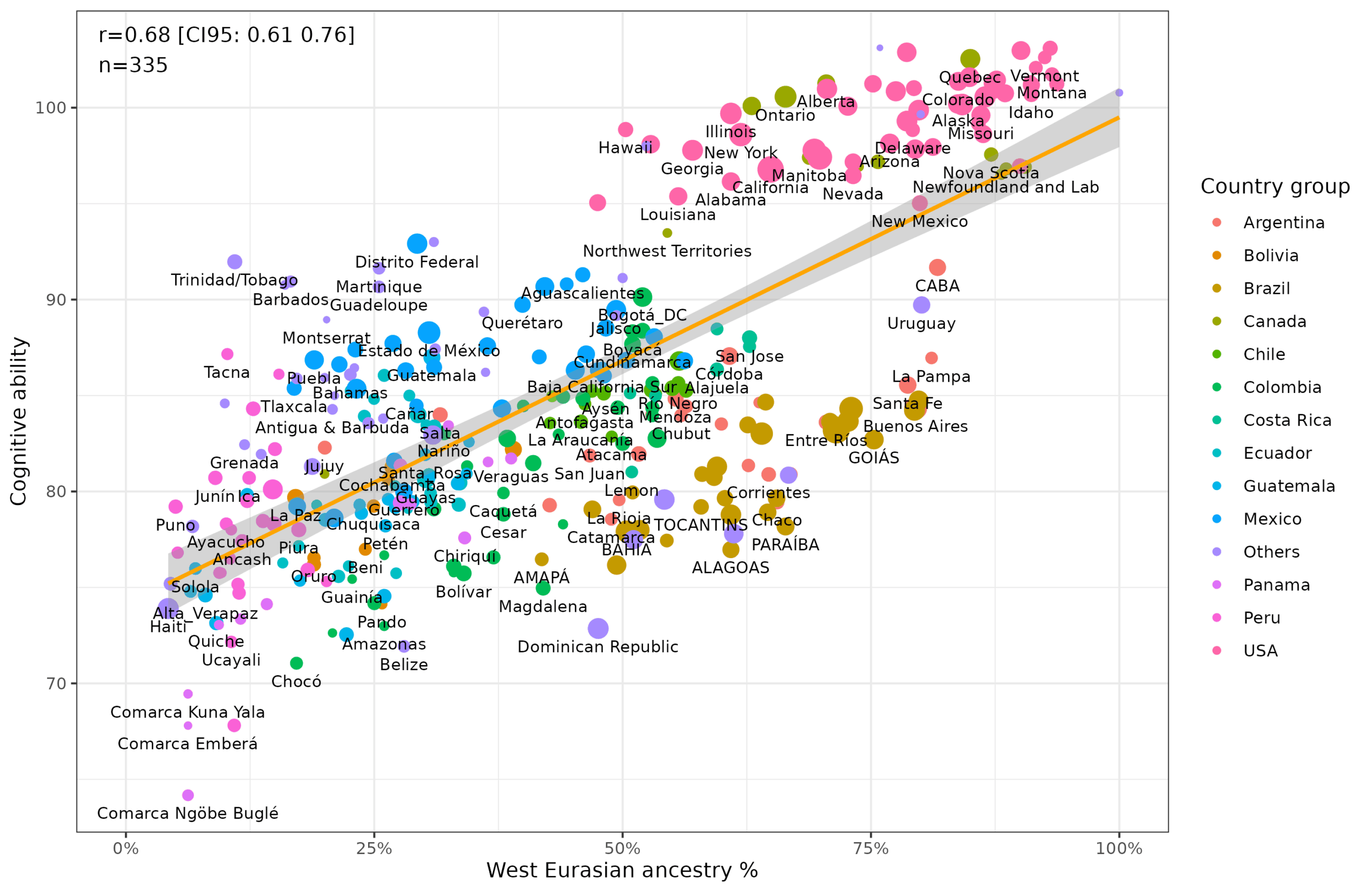
We are finally ready to share our big follow-up to the first Admixture in the Americas project from 2016. Fuerst, J., & Kirkegaard, E. O. W. (2025, December 5). Continental…
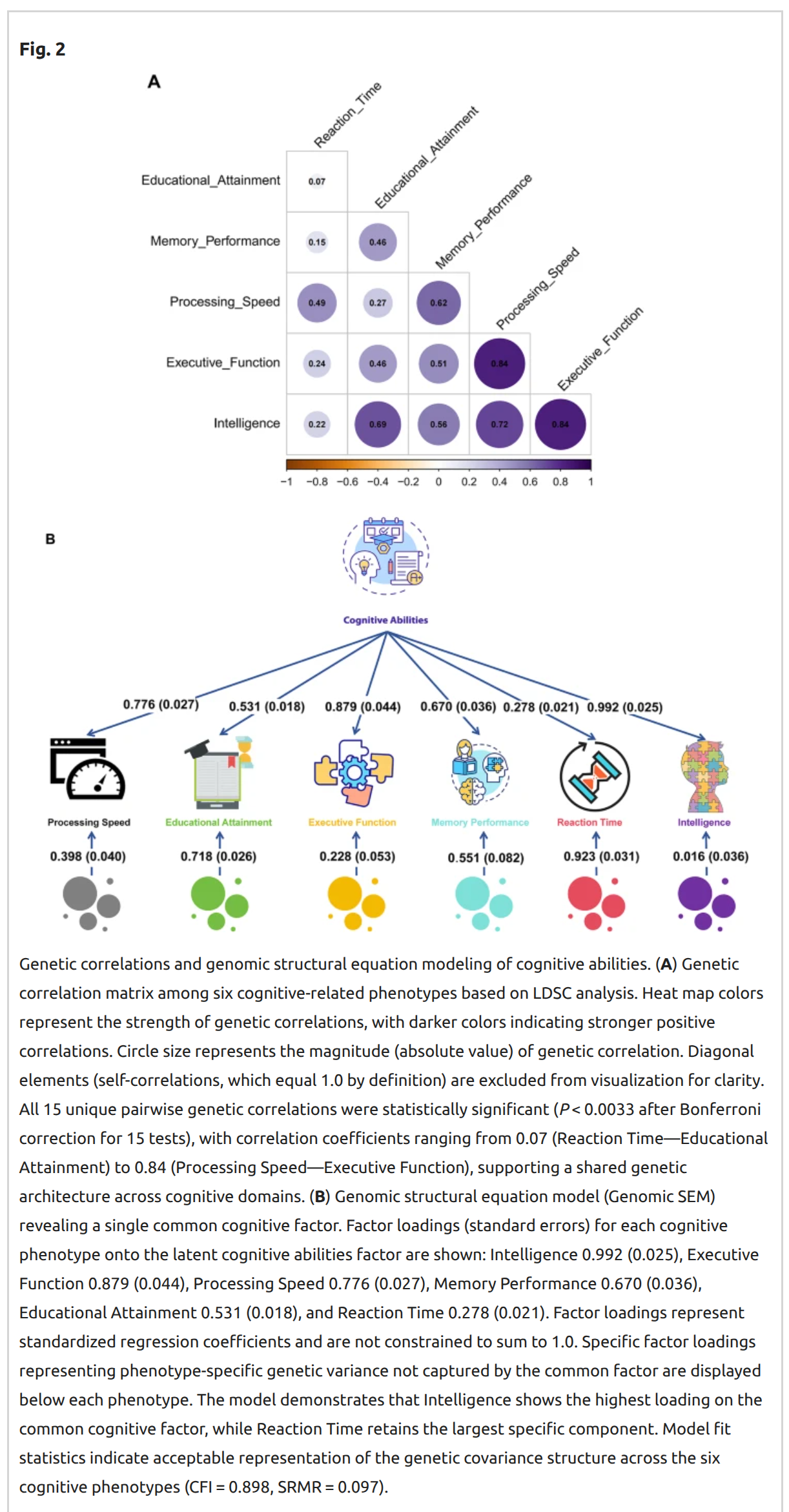
There's a new genetic analysis of intelligence out: Chen, H., Liao, Y., Tang, L., Wei, X., Li, T., & Chen, W. (2025). Multivariate genome-wide analysis reveals shared genetic architecture and…
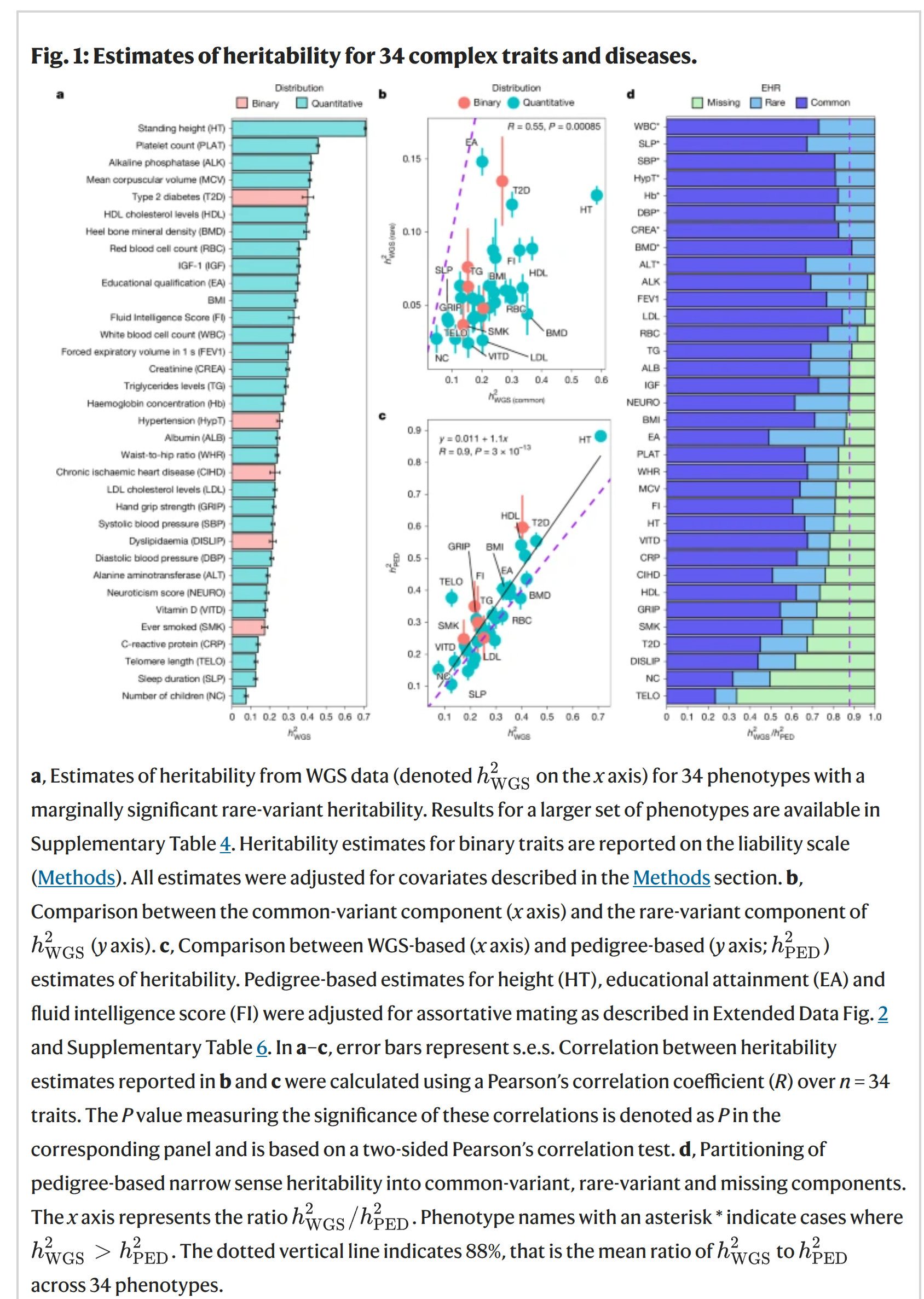
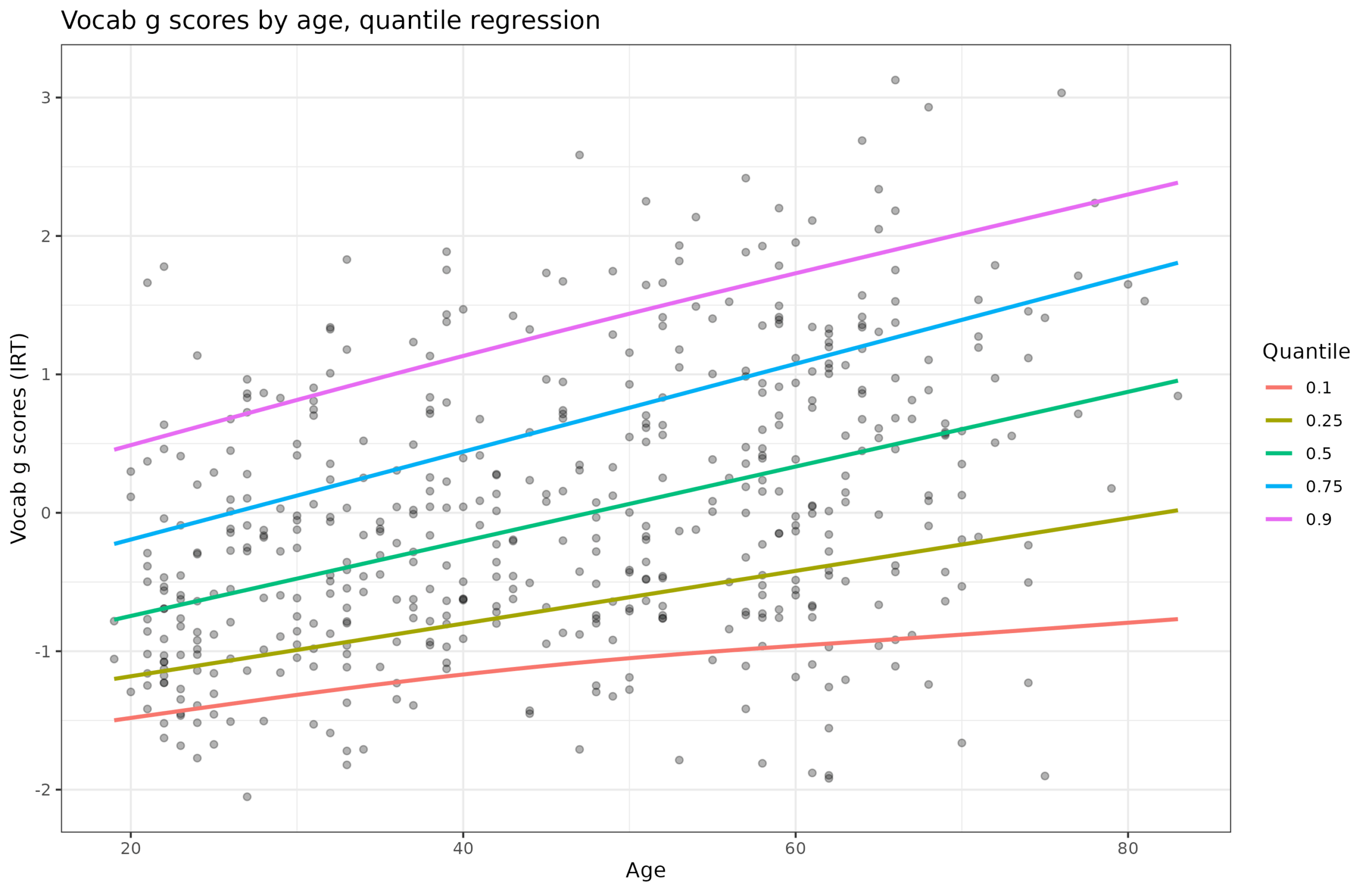
I wasn't going to blog this, since I prefer not covering the same ground as others, but due to multiple requests and questions, I will cover it anyway. It's this…
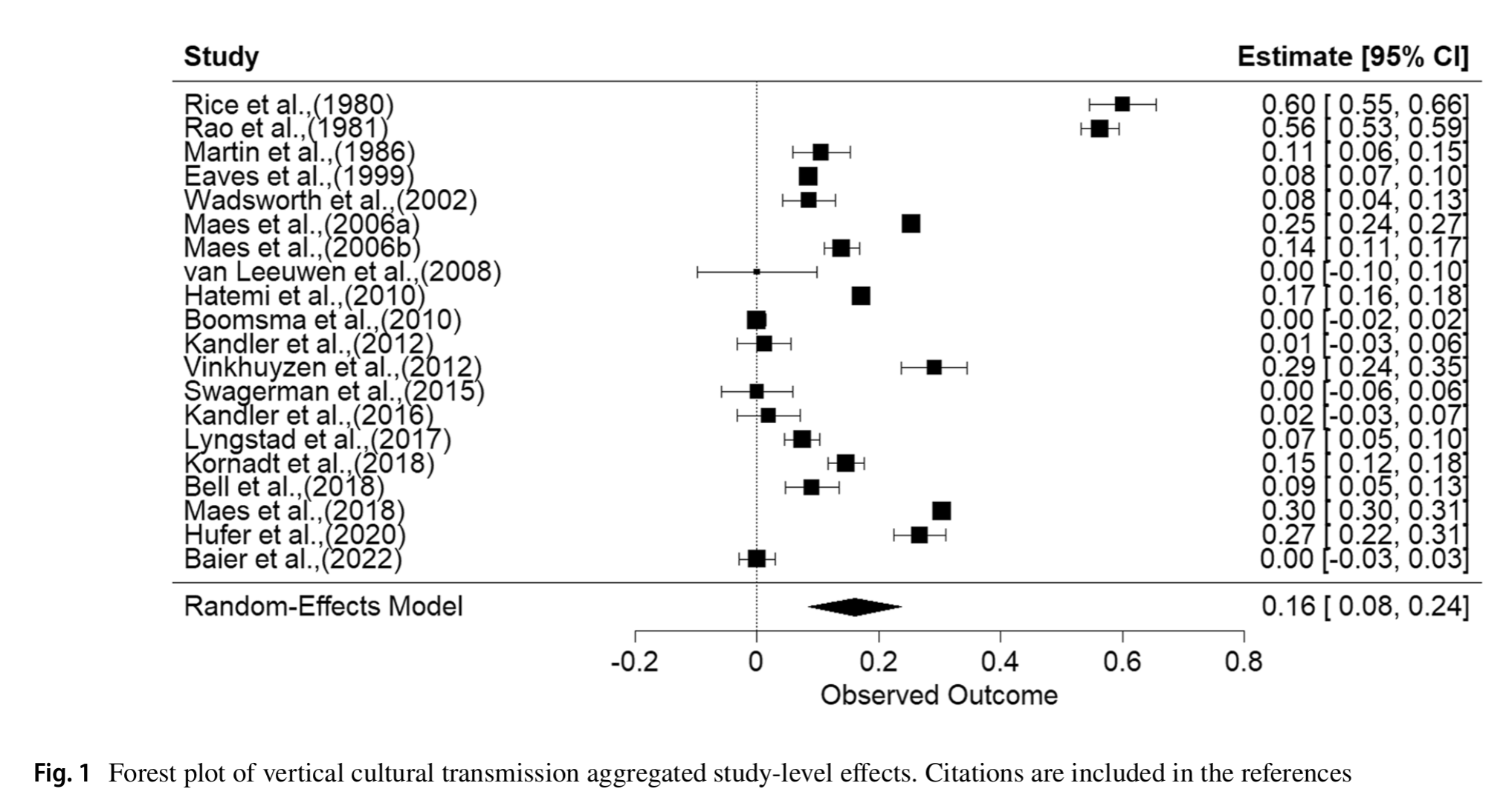
Last year Marcus Feldman and Kevin Lala published one of those anti-hereditarian pieces: Lala, K. N., & Feldman, M. W. (2024). Genes, culture, and scientific racism. Proceedings of the National…
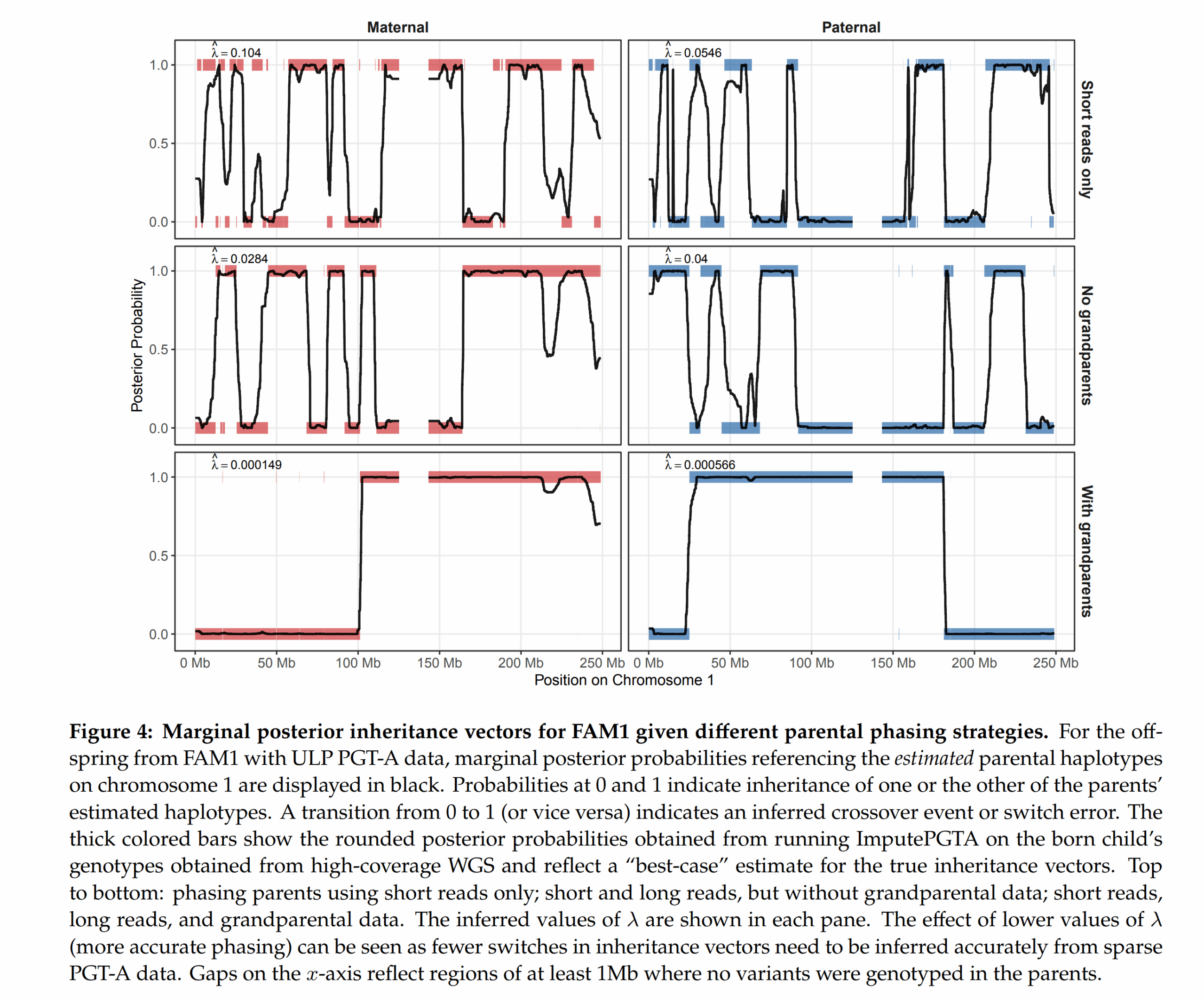
Things are moving fast in the embryo selection sector. There was never really any technological barriers here the last 10+ years, just the usual social taboos and various incompetent people…
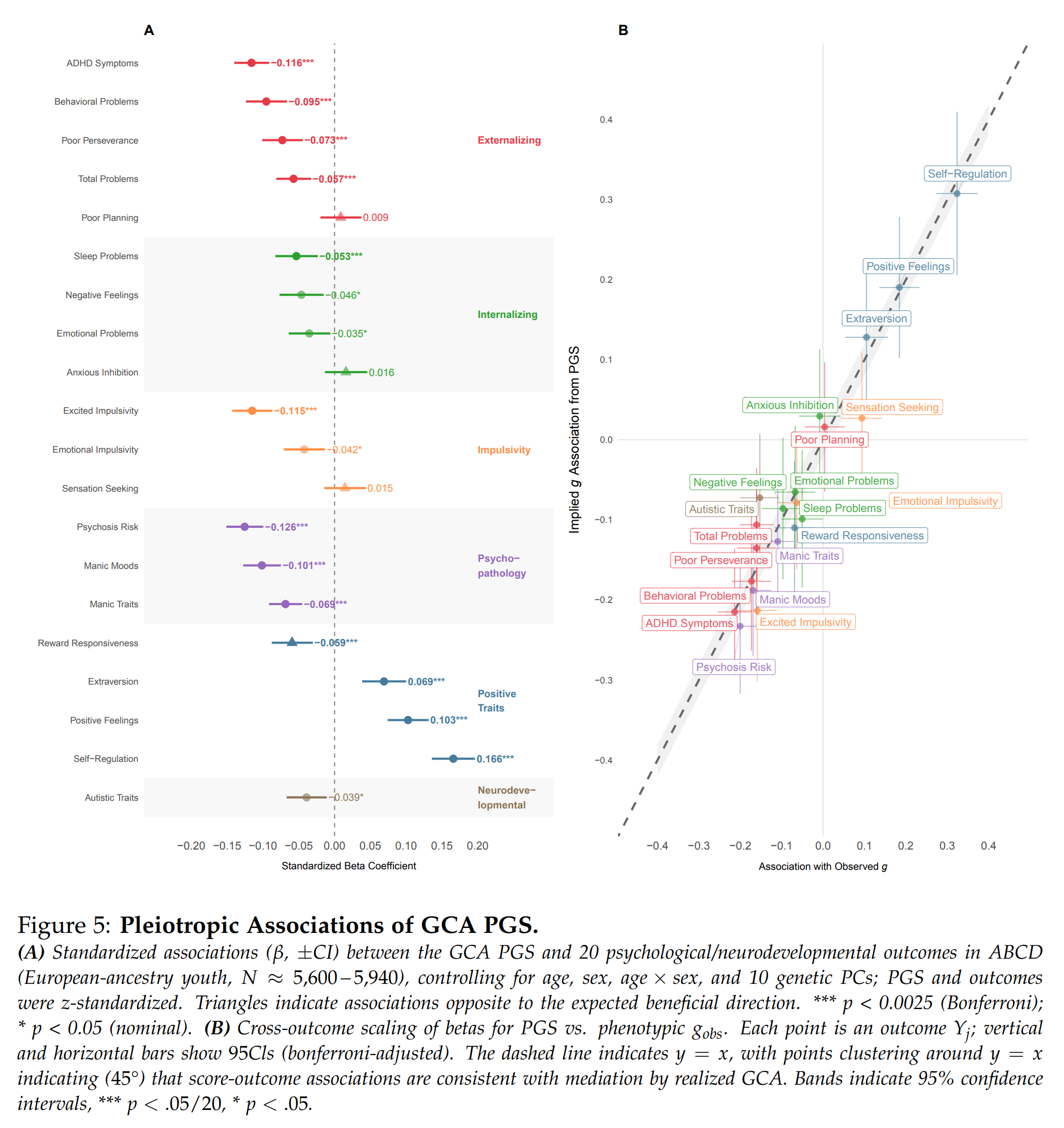
There's two exciting news today in embryo selection. The technology is finally moving forward at an appreciable rate. Maybe we are not dysgenics doomed after all. First off, Herasight --…
Suppose you have created a new scale of something ("Science Blog Rating Scale"). It can be psychology or some other soft science, but it doesn't have to be. Suppose you…