I recently had my full genome sequenced at Nebula genomics for their 30x. The results were satisfactory:

Good eyes too:
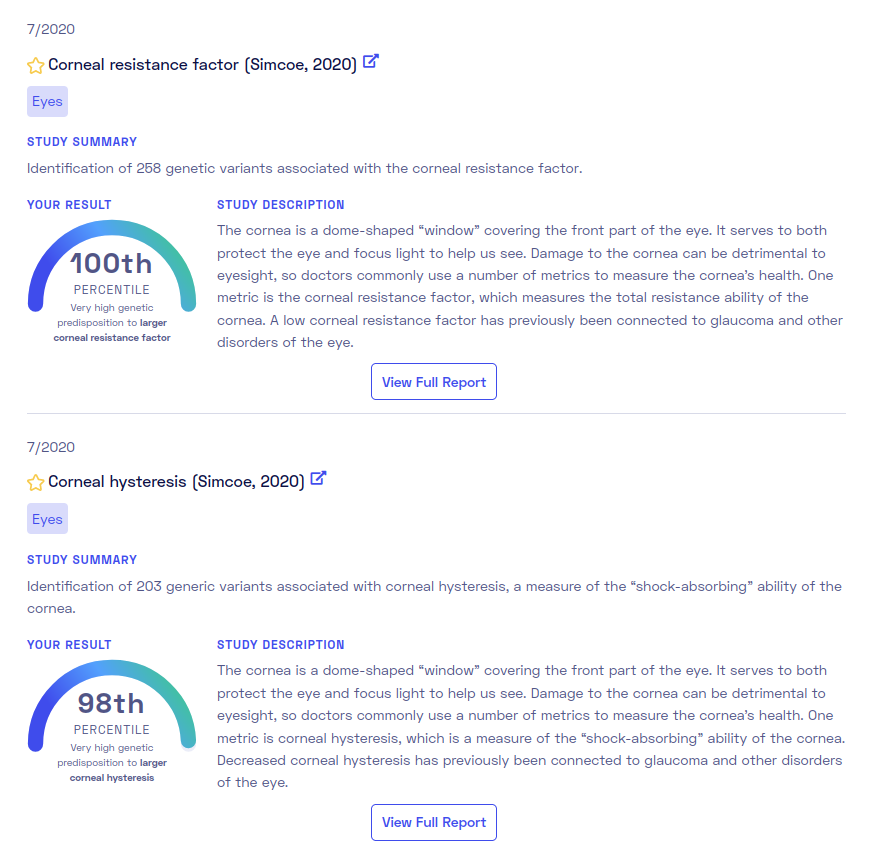
Literally unbreakable:

You ready to get your own great results? Too good to be true? Of course it is. I just browsed their findings and picked some implausible ones I liked. Most of their model predictions are clearly useless as they are based on tiny numbers of variants. Furthermore, they don’t really report the model accuracies. It doesn’t matter if one is in the 100th centile of a model that doesn’t work. So let’s look closer at the testosterone model. Here’s the testosterone report: Testosterone level (Ruth, 2020)
Testosterone is the main male sex hormone. However, it regulates bodily functions, like muscle development and fertility, in both sexes. This study examined over 425,000 individuals of European ancestry from the UK Biobank database to identify genetic factors associated with testosterone level. The researchers linked over 200 genetic variants to testosterone level in both sexes. These variants explained 17% of the heritability of testosterone level in men and 13% of the heritability in women. The study also found differences between sexes with some variants showing opposite effect directions. Furthermore, the results of this study indicate that genetic predisposition to higher testosterone is beneficial to men but harmful to women. For example, genetically determined higher testosterone level in men decreased the risk of type 2 diabetes but increased that risk in women.
It looks good. It’s based on a GWAS of the UKBB, n = 425,000. but I don’t for a moment think I’m actually in the 100th centile. The reported variance explained %’s are false. These are not the polygenic score heritabilities, but the SNP heritabilities (GREML type). The GWAS authors don’t appear to give the variance % for the polygenic scores, but they do give these plots:
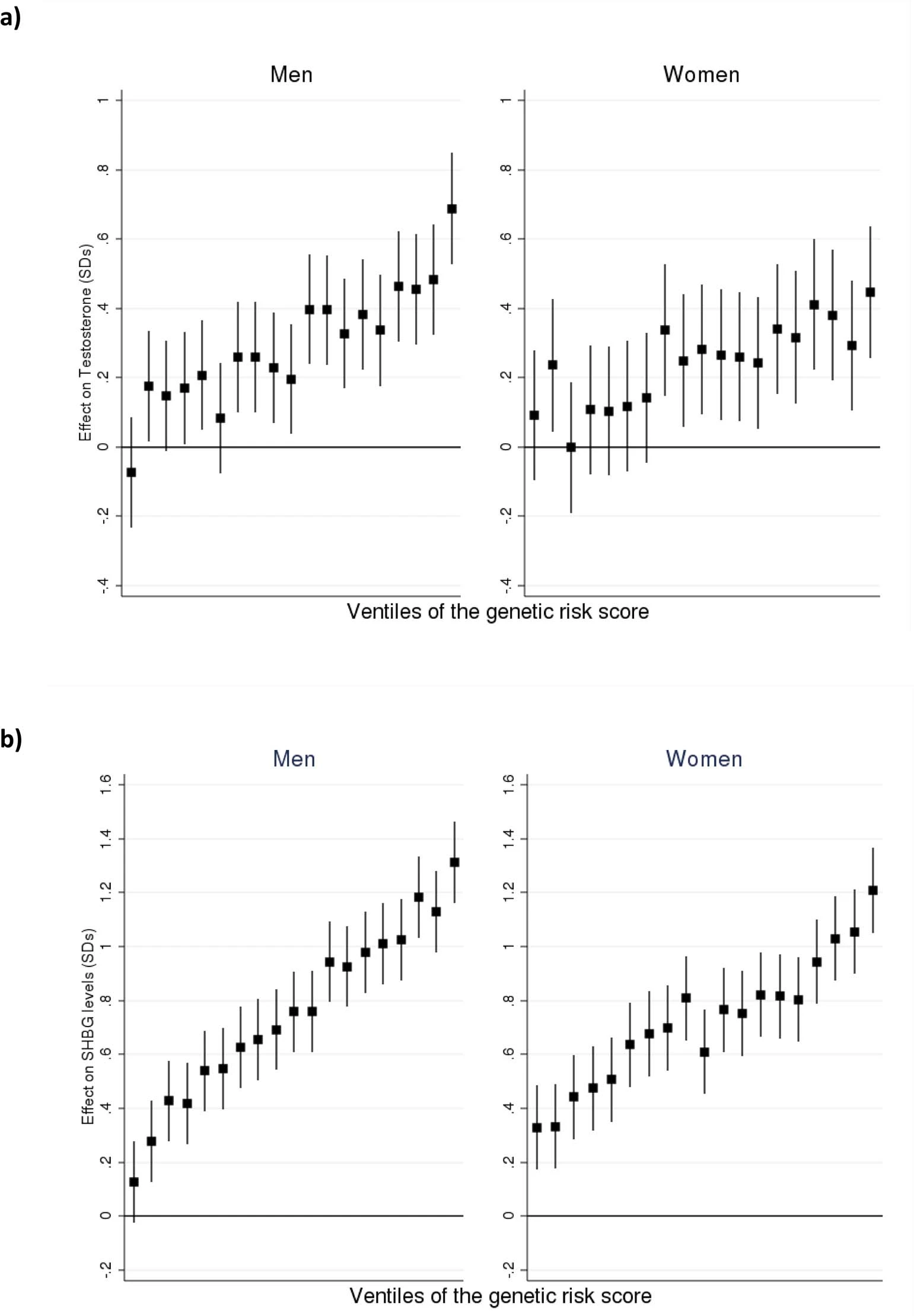
So, the top 5% polygenic scores appear to be about 0.75 d higher in T than the bottom 5%, in men. For women, top 5% vs. bottom 5% looks like 0.35 d. I reverse engineered these gaps and figured out that this corresponds to correlations of about .18 and .08. Not too bad for a first try GWAS. But also note that their top and bottom 5% groups were outliers for men, so the effect size is probably overestimated a here. I don’t know what Nebula messed up in their coding, but a lot of their models produce extreme centiles for me. My best guess is that they implemented the models incorrectly, either by incorrect scoring or by lack of proper quality control.
More disappointing was that Nebula doesn’t supply premade structural variant and copy number variant calls. These are the kinds of non-SNP genetic variants that you keep hearing about. The most famous in intelligence research is DUF1220, which I wrote about before. These kinds of variants can’t be measured directly using the array technology, and can only sometimes be imputed accurately. So the use of high coverage whole genome sequencing (WGS) should mean we can finally look at these variants. But then they fail at the last step of giving users premade results for browsing. Weird decision! Here’s the files they offer for download:
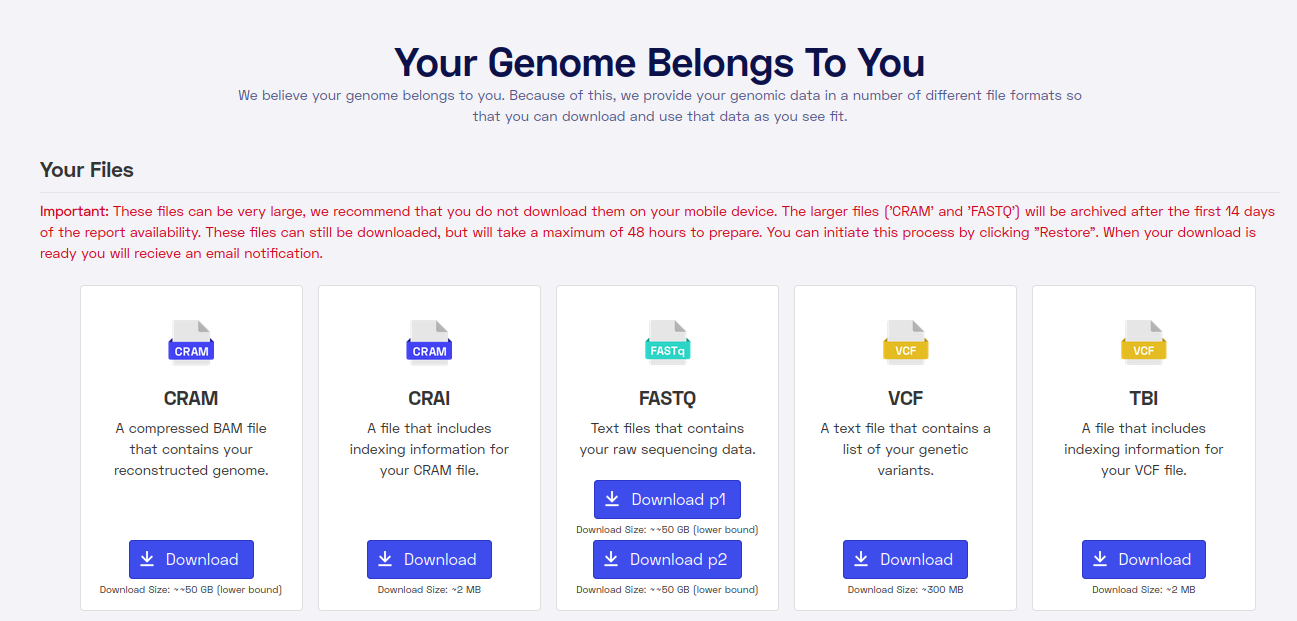
In fact, you can download my data right here. The raw datafile (CRAM) is 56GB of letters they read of my genome.
My array (SNP) genome has been public for about 9 years already, so I don’t think I am taking a big step in making public my full genome. Hopefully, some readers will have a look at this and maybe you can find some weird disease I have. Hopefully, you won’t find some weird criminal in my extended family!
What else does Nebula offer? Well, first off, they were extremely slow. It took months to get my results. Second, they offer some quite terrible paid ancestry results:
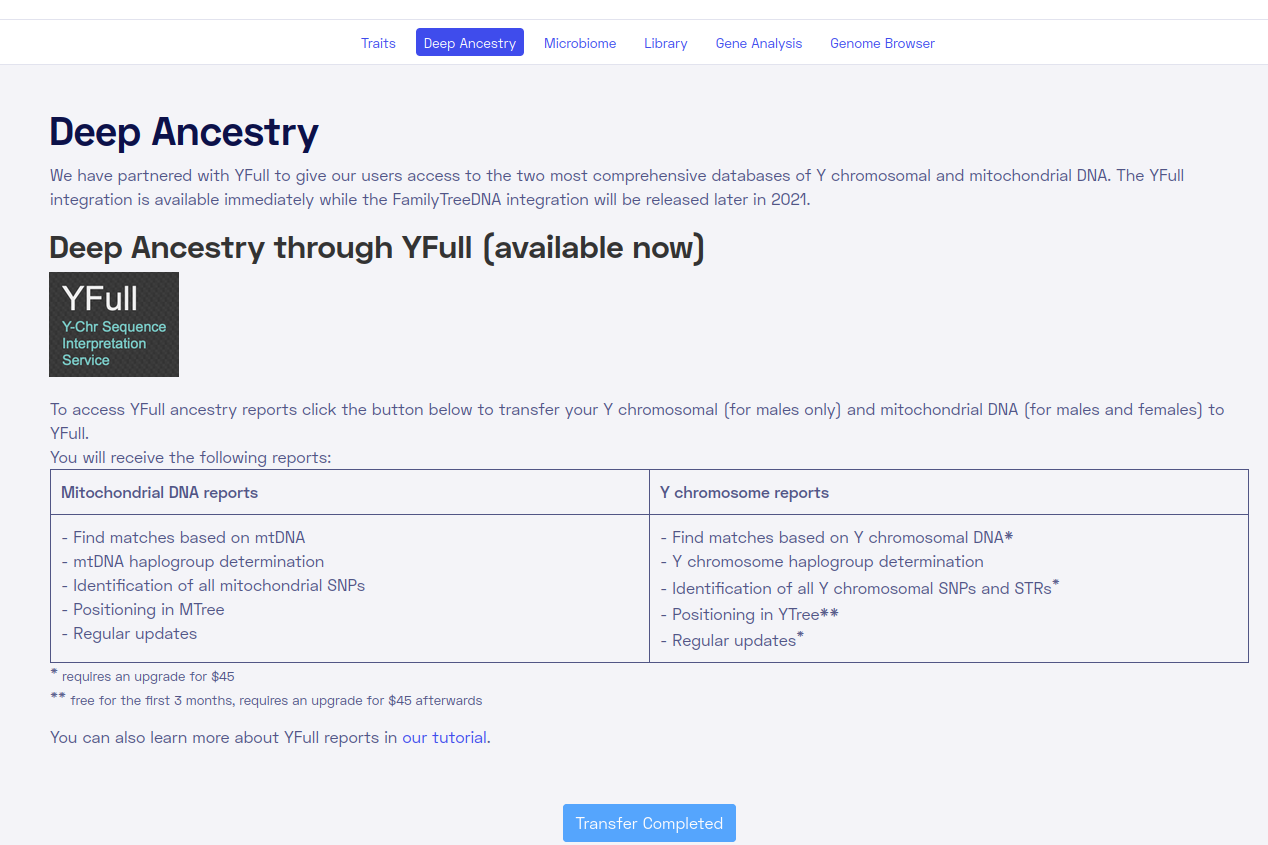
Who wants to pay 45 USD to read about some Y/MT results? These are included in 23andme and every other service and are very easy to do, and not very interesting. Nebula doesn’t even offer any global ancestry results. They could have easily collected some public genomes and at least provided people with people with European%, Asian% or something, but no. Again, odd.
In terms of data collection, they also go the survey route of 23andme:
So if they keep not going bankrupt (I hear they are doing poorly), there should be some GWASs based on WGS data that use Nebula data. Well, so I hope!
What about data quality? Well, here’s how their SNP and indel calls look like (bcftools view):
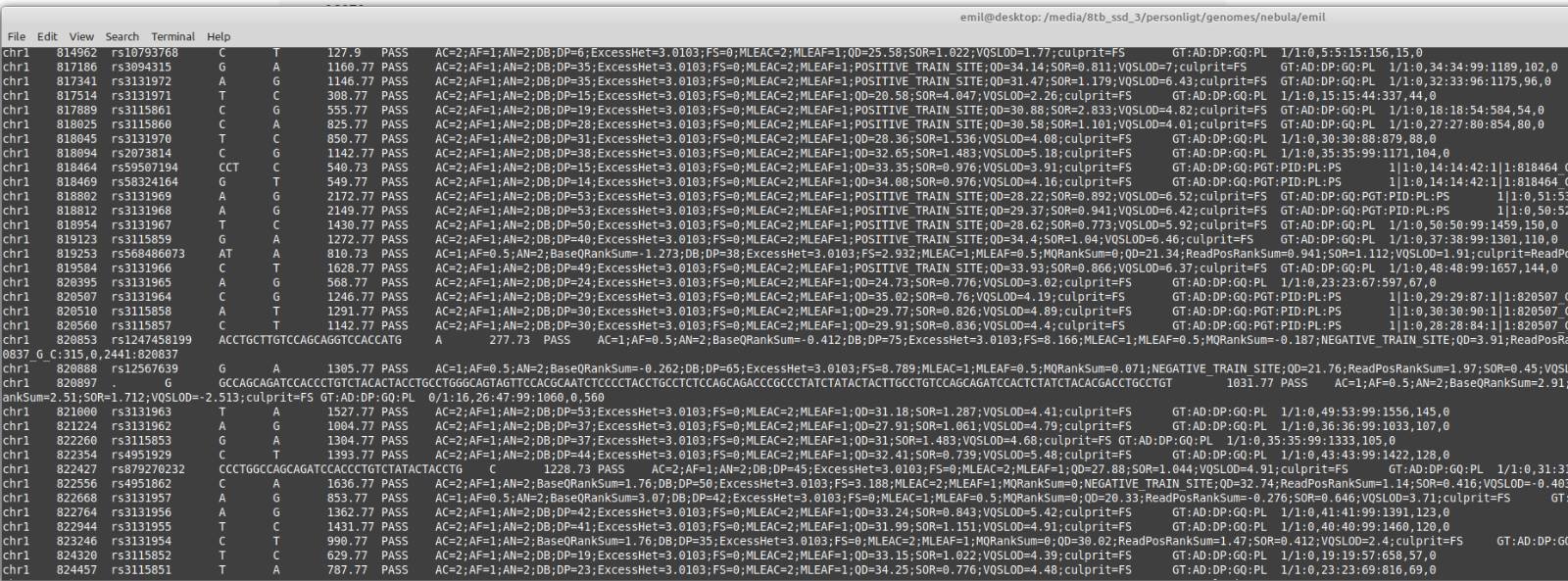
The data appears to be in hg38 build, and they have helpfully added the rsid’s. That means we can easily compare the results to the array data from 23andme. I picked a random the first SNP, rs3094315. 23andme says I have AA. Nebula provides a genome browser, which shows this plot:

It’s somewhat confusing at first, but actually it shows each read of this region and which letter it saw. Since this is 30x coverage, there should be about 30 reads, and indeed, there are 34. All of these were A’s, so clearly I must be AA at this loci. Hooray, n=1 agreement! Their VCF file looks like this (in R). Here’s the same SNP, line 1:
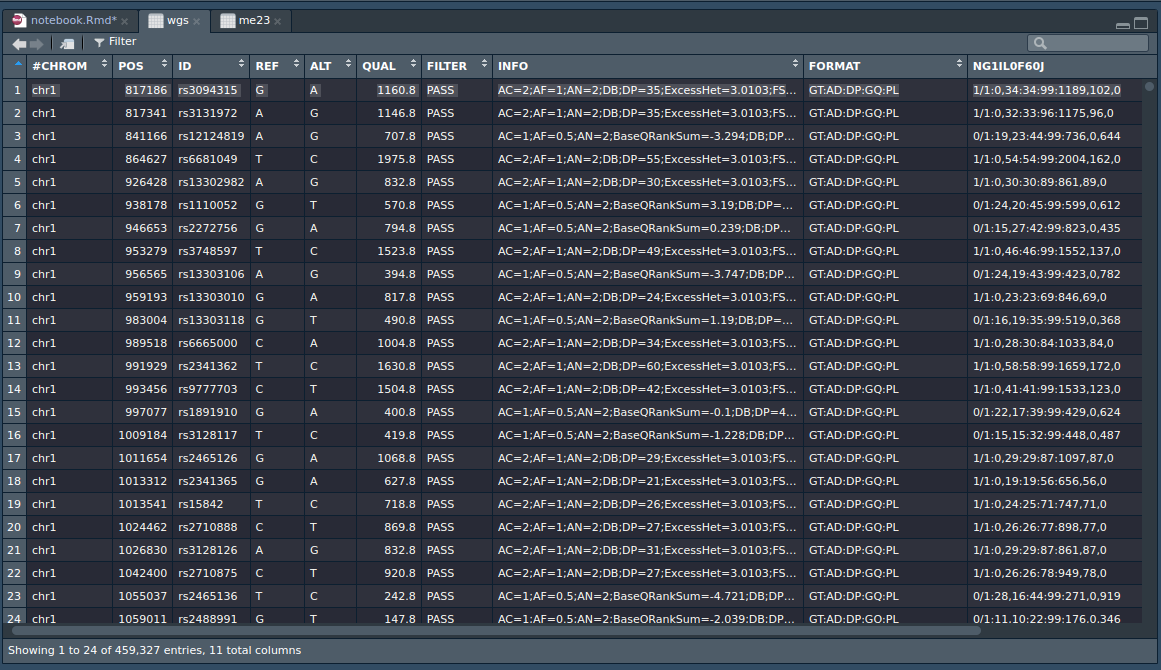
The data are in the last column (VCF files are transposed basically, metadata in rows, data in columns). The GT field is the genotype, so it should show that I have two ALT alleles. It says 1/1 which indeed means I have 2 copies of the second allele (first in ALT column), which is A. In fact, their VCF file doesn’t include the loci where you have the most common genotype (i.e. REF REF), presumably to save a lot of space as otherwise the VCF file would be huge of redundant information. All human genomes are identical at most loci, there isn’t any variation. Of the ones that show some variation at least, most people still have the most common alleles, so noting this in a file would be a waste of time. Anyway, if one is trying to score genomes, then, one has to fill in these missing variants.
All in all, WGS data is not very useful for end-users as of yet because most genetic prediction models (‘GWASs’) aren’t based on WGS data, so there’s not much to use the extra information for. I expect this to change in the coming years. In terms of utility of getting WGS done now, then, there isn’t much. If you wait, you will get the same done cheaper and at higher quality. If you have a family member who is dying or very old, it is sensible to get their WGS done now before it is too late. For those who aren’t dying, WGS now is really only for enthusiasts who want to dig into potential weird rare variants, including the structural ones. It is therefore a pity that they don’t provide calls for these by default!
