There’s a highly interesting new paper out:
(Mendelians are also known as monogenic disorders because they are inherited in patterns that follow Mendel’s laws.)
Abstract:
Discovering the genetic basis of a Mendelian phenotype establishes a causal link between genotype and phenotype, making possible carrier and population screening and direct diagnosis. Such discoveries also contribute to our knowledge of gene function, gene regulation, development, and biological mechanisms that can be used for developing new therapeutics. As of February 2015, 2,937 genes underlying 4,163 Mendelian phenotypes have been discovered, but the genes underlying ∼50% (i.e., 3,152) of all known Mendelian phenotypes are still unknown, and many more Mendelian conditions have yet to be recognized. This is a formidable gap in biomedical knowledge. Accordingly, in December 2011, the NIH established the Centers for Mendelian Genomics (CMGs) to provide the collaborative framework and infrastructure necessary for undertaking large-scale whole-exome sequencing and discovery of the genetic variants responsible for Mendelian phenotypes. In partnership with 529 investigators from 261 institutions in 36 countries, the CMGs assessed 18,863 samples from 8,838 families representing 579 known and 470 novel Mendelian phenotypes as of January 2015. This collaborative effort has identified 956 genes, including 375 not previously associated with human health, that underlie a Mendelian phenotype. These results provide insight into study design and analytical strategies, identify novel mechanisms of disease, and reveal the extensive clinical variability of Mendelian phenotypes. Discovering the gene underlying every Mendelian phenotype will require tackling challenges such as worldwide ascertainment and phenotypic characterization of families affected by Mendelian conditions, improvement in sequencing and analytical techniques, and pervasive sharing of phenotypic and genomic data among researchers, clinicians, and families.
The issue is worth discussing in a bit of detail, because there are important implications for multiple important areas. First, the sheer scale:
Much remains to be learned. The HGP and subsequent annotation efforts have established that there are ∼19,000 predicted protein-coding genes in humans.9, 10 Nearly all are conserved across the vertebrate lineage and are highly conserved since the origin of mammals ∼150–200 million years ago,11, 12, 13 suggesting that certain mutations in every non-redundant gene will have phenotypic consequences, either constitutively or in response to specific environmental challenges. The continuing pace of discovery of new Mendelian phenotypes and the variants and genes underlying them supports this contention.
Humans have about 19k protein coding regions (which I will call genes here) and these seem to be high preserved over time, meaning that they are very likely to be functional in some important way. It’s possible that a large number of them are critical to life, meaning that any major disorder in them will inevitably cause spontaneous abortion (a common thing for serious disorders, e.g. 99% of Turner syndrome embryos get aborted automatically) or stillbirth. In a sense, then, these genetic disorders all share a few deadly phenotypes, and it will not be possible to find any phenotypes for these genes among the living. This is based on the strong assumption of total loss of function for the mutations, which is not realistic. The authors note that we have animal evidence:
Studies in mice with engineered loss-of-function (LOF) mutations suggest that the majority of the gene knockouts compatible with survival to birth are associated with a recognizable phenotype, whereas ∼30% of gene knockouts lead to in utero or perinatal lethality.50 Of the latter, it remains to be determined whether partial LOF mutations (i.e., hypomorphic alleles) or other classes of mutations (e.g., gain of function, dosage differences due to gene amplification,51 etc.) in the same genes might result in viable phenotypes. Nevertheless, given the high degree of evolutionary conservation between humans and mice, mutations in the majority of non-redundant human protein-coding genes are likely to result in Mendelian phenotypes, most of which remain to be characterized (Figure 2).
While many mutations are large deletions that cause total loss of function, many others are smaller deletions, duplications (e.g. Huntington’s) or alternative versions (SNPs etc.; e.g. thalassemia). There can be multiple disorders per gene because there can be different mutations that cause different problems, e.g. cause a protein to fold in two different, but both problematic ways.
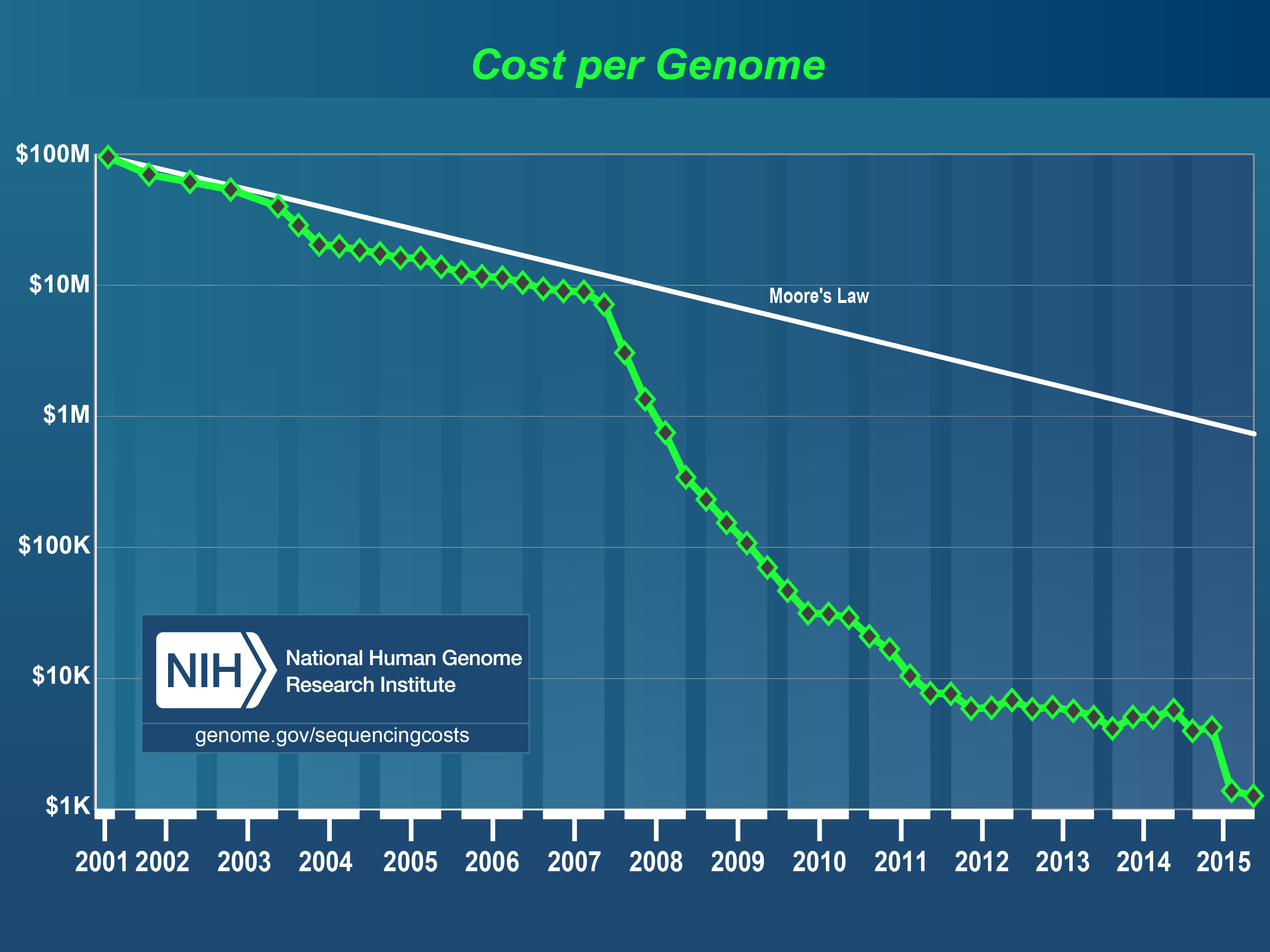
Overall, my takeaway is that 1) there are likely a very large number of Mendelians yet to be discovered among the living, 2) a substantial fraction of genes currently not associated with any disorder are critical for life i.e. cause death to fetus hence there is no phenotype to find aside from that (30% perhaps), 3) it will be hard to find many of the disorders that have relatively mild effects both because of the inherent needle in haystack situation, and because of measurement problems, especially with regards to copy number variants (different numbers of a given sequence of DNA). Proper measurement of complex genetic variation (i.e. non-SNP) will probably require both improved mathematical modeling (I’m looking at you, sparsity/penalization) as well as sequencing. Sequencing is still terrible in a cost-benefit sense, but array data can get us a lot further until sequencing becomes price competitive.
The burden of disease
In aggregate, clinically recognized Mendelian phenotypes compose a substantial fraction (∼0.4% of live births) of known human diseases, and if all congenital anomalies are included, ∼8% of live births have a genetic disorder recognizable by early adulthood.27 This translates to approximately eight million children born worldwide each year with a “serious” genetic condition, defined as a condition that is life threatening or has the potential to result in disability.28 In the US alone, Mendelian disorders collectively affect more than 25 million people and are associated with high morbidity, mortality, and economic burden in both pediatric and adult populations.28,29 Birth defects, of which Mendelian phenotypes compose an unknown but most likely substantial proportion, are the most common cause of death in the first year of life, and each year, more than three million children under the age of 5 years die from a birth defect, and a similar number survive with substantial morbidity. Beyond the emotional burden, each child with a genetic disorder has been estimated to cost the healthcare system a total of $5,000,000 during their lifetime.28,29
It remains a challenge to diagnose many Mendelian phenotypes by phenotypic features and conventional diagnostic testing. In a general clinical genetics setting, the diagnostic rate is ∼50%.30 Across a broader range of rare diseases, diagnostic rates are even lower. For example, in the NIH Undiagnosed Disease Program, the diagnostic rate was, despite state-of-the-art evaluations, 34% in adults and 11% in children.31 Moreover, the time to diagnosis is often prolonged (the “diagnostic odyssey”); in a European survey of the time to diagnosis of eight rare diseases, including cystic fibrosis (MIM: 602421) and fragile X syndrome (MIM: 309550), 25% of families waited between 5 and 30 years for a diagnosis, and the initial diagnosis was incorrect in 40% of these families.32
There are substantial gains to be made from cleaning out these disorders from our common human gene pool. I see no reason to expect rigid Mendelian causation (complete dominance), meaning that heterozygous copies of poor variants likely also cause some milder problems, which are then often missed due to the usual statistical and clinical problems. If we are unlucky, there will be many variants that show heterozygote superiority (such as the sickle cell variant for malaria resistance). I say unlucky because heterozygosity is brittle and would need constant management of reproduction to sustain. If both parents are heterozygous (AB, AB), 50% of their children will be too (AB, BA), but the other 50% will be split between the two homozygous genotypes (AA, BB).
Intervention relevance
Development of new therapeutics to address common diseases that constitute major public-health problems is limited by the ignorance regarding the fundamental biology underlying disease pathogenesis.60 As a consequence, 90% of drugs entering human clinical trials fail, commonly because of a lack of efficacy and/or unanticipated mechanism-based adverse effects.61 Studies of families affected by rare Mendelian phenotypes segregating with large-effect mutations that increase or decrease risk for common disease can directly establish the causal relationship between genes and pathways and common diseases and identify targets likely to have large beneficial effects and fewer mechanism-based adverse effects when manipulated. For example, certain Mendelian forms of high and low blood pressure are due to mutations that cause increases and decreases, respectively, in renal salt reabsorption and net salt balance; these discoveries identified promising new therapeutic targets, such as KCNJ1 (potassium channel, inwardly rectifying, subfamily J, member 1 [MIM: 600359]), for which drugs are now in clinical trials. Understanding the role of salt balance in blood pressure has provided the scientific basis for public-health efforts in more than 30 countries to reduce heart attacks, strokes, and mortality by modest reduction in dietary salt intake.62 Similarly, understanding the physiological effects of CFTR (cystic fibrosis transmembrane conductance regulator [MIM: 602421]) mutations responsible for cystic fibrosis has led to allele-specific therapies that significantly improve pulmonary function in affected individuals.63 Other common-disease drugs based on gene discoveries for Mendelian phenotypes (e.g., orexin antagonists for sleep,64 beta-site APP-cleaving enzyme 1 [BACE1] inhibitors for Alzheimer dementia,65 proprotein convertase, subtilisin/kexin type 9 [PCSK9] monoclonal antibodies to lower low-density lipoprotein levels66) are undergoing advanced clinical trials. Discoveries such as these will directly facilitate the goals of the Precision Medicine Initiative.67
This is an instance of the more general pattern: very complex systems are really hard to understand from first principles. However, by carefully observing existing variation in them, it is possible to make (more) plausible and testable hypotheses about the causal pathways that can be used for interventions. It seems very likely that improved knowledge of Mendelian genetics would allow us to propose and test hypotheses that have higher prior probabilities, which in the end lead us to more easily identify working interventions. To put it metaphorically, there are too many places to dig for gold (=very low, uniform prior landscape), but by using existing genetic variation, we can figure out where it is promising to dig (=more spiky landscape).
If we want to get serious
If we want to get serious about finding important genetic variation, we should genotype all humans for which we have reasonable phenotype data. The Nordic countries are the perfect place to begin because these are extremely rich (despite recent dysfunctional immigration) and importantly, they have very comprehensive ‘social security number’-like systems that cover everybody in the country and is linked to virtually all other public databases, including medical. This means that every diagnosis and every (prescription) treatment are already in the same linkable database. Researchers are beginning to use these data, but they are unfortunately taking a candidate-gene-like approach by only testing for specific relationships, instead of taking a phenome-‘treatome’ wide approach akin to GWASs. Still, they produce interesting studies such as:
Antipsychotics, mood stabilisers, and risk of violent crime
Methods
We used linked Swedish national registers to study 82 647 patients who were prescribed antipsychotics or mood stabilisers, their psychiatric diagnoses, and subsequent criminal convictions in 2006–09. We did within-individual analyses to compare the rate of violent criminality during the time that patients were prescribed these medications versus the rate for the same patients while they were not receiving the drugs to adjust for all confounders that remained constant within each participant during follow-up. The primary outcome was the occurrence of violent crime, according to Sweden’s national crime register.
Findings
In 2006–09, 40 937 men in Sweden were prescribed antipsychotics or mood stabilisers, of whom 2657 (6·5%) were convicted of a violent crime during the study period. In the same period, 41 710 women were prescribed these drugs, of whom 604 (1·4 %) had convictions for violent crime. Compared with periods when participants were not on medication, violent crime fell by 45% in patients receiving antipsychotics (hazard ratio [HR] 0·55, 95% CI 0·47–0·64) and by 24% in patients prescribed mood stabilisers (0·76, 0·62–0·93). However, we identified potentially important differences by diagnosis—mood stabilisers were associated with a reduced rate of violent crime only in patients with bipolar disorder. The rate of violence reduction for antipsychotics remained between 22% and 29% in sensitivity analyses that used different outcomes (any crime, drug-related crime, less severe crime, and violent arrest), and was stronger in patients who were prescribed higher drug doses than in those prescribed low doses. Notable reductions in violent crime were also recorded for depot medication (HR adjusted for concomitant oral medications 0·60, 95% CI 0·39–0·92).
Medication for Attention Deficit–Hyperactivity Disorder and Criminality
Methods
Using Swedish national registers, we gathered information on 25,656 patients with a diagnosis of ADHD, their pharmacologic treatment, and subsequent criminal convictions in Sweden from 2006 through 2009. We used stratified Cox regression analyses to compare the rate of criminality while the patients were receiving ADHD medication, as compared with the rate for the same patients while not receiving medication.Results
As compared with nonmedication periods, among patients receiving ADHD medication, there was a significant reduction of 32% in the criminality rate for men (adjusted hazard ratio, 0.68; 95% confidence interval [CI], 0.63 to 0.73) and 41% for women (hazard ratio, 0.59; 95% CI, 0.50 to 0.70). The rate reduction remained between 17% and 46% in sensitivity analyses among men, with factors that included different types of drugs (e.g., stimulant vs. nonstimulant) and outcomes (e.g., type of crime).
These studies are not as good as RCTs because while they control for any kind of confounder that’s stable across the lifespan of a person (including genetics and upbringing), they do not control for time-variant confounders. The most obvious one here is that of reverse causality from good and bad period/mood fluctuation that cause people both to act foolishly and stop taking drugs. I know some schizophrenics and this seems plausible to me to some extent.
If we really, really want to get serious, we want to start randomizing treatment across the entire health system. This can done more easily when one has publicly paid health care (take that capitalism!). Here I don’t mean randomizing between placebo and actual treatments (placebo doesn’t work for non-subjective problems), I mean randomizing patients between different treatments for the same problem. In fact, treatment comparison studies are rarely conducted, so we often don’t actually know which treatment is better (see e.g. discussion of MPD vs. Amphetamine for ADHD). Essentially, the way doctors currently work, is that when they have a specific condition they want to treat, they semi-randomly, maybe in conjunction with the patient, choose a treatment plan. Instead, they could ask the patient whether they want to take part in the nation-wide experiment for the good of all (maybe we can give such patients a bumper sticker to use for social signaling just like we do for blood and organ donors now). Many will consent to this, and the doctor will then ask the computer which treatment the patient should get. The computer picks one at random, saves this info in the database, and things move on as usual. This way, we can automatically collect very large datasets of randomized data for any condition where we have multiple approved treatments and we’re unsure which is better. With the added genomic data, we can do pharmacogenomics too (i.e. look for genotype x treatment interactions).