There are many studies that look at signs for selection using large genomic datasets. This post provides a selective summary of such studies from 2018 to now, so very recent progress. There are in general two ways to approach the question of selection. First, one can look at known variants for traits found via GWASs, and look for evidence of selection. Second, one can look at all variants in the genome and look for evidence of selection, and among those found, try to determine which traits they relate to, either by comparing with GWAS results or by relations to known gene functions or pathways.
The studies are given in approximate chronological order. The list does not include Davide Piffer’s studies, since these are spread out in many preprints and are not so organized. It is easier to find them via his Google Scholar. Likewise, does not include the various admixture/ancestry based results, including my own work. This list is mainly about quantitative/population genetics/genomics of selection, not other means of determining genetic causes of group gaps (though these would have arisen by either divergent selection or drift).
Studies
- Guo, J., Yang, J., & Visscher, P. M. (2018). Leveraging GWAS for complex traits to detect signatures of natural selection in humans. Current Opinion in Genetics & Development, 53, 9-14.
Natural selection can shape the genetic architecture of complex traits. In human populations, signals of positive selection at genetic loci have been detected through a variety of genome-wide scanning approaches without the knowledge of how genes affect traits or fitness. In the past decade, genome-wide association studies (GWAS) have provided unprecedented insights into the genetic basis of quantitative variation in complex traits. Summary statistics generated from these GWAS have been shown to be an extraordinary data source that can be utilized to detect and quantify natural selection in the genetic architecture of complex traits. In this review, we focus on recent discoveries about selection on genetic variants associated with human complex traits based on GWAS-facilitated methods.
- Racimo, F., Berg, J. J., & Pickrell, J. K. (2018). Detecting polygenic adaptation in admixture graphs. Genetics, 208(4), 1565-1584.
An open question in human evolution is the importance of polygenic adaptation: adaptive changes in the mean of a multifactorial trait due to shifts in allele frequencies across many loci. In recent years, several methods have been developed to detect polygenic adaptation using loci identified in genome-wide association studies (GWAS). Though powerful, these methods suffer from limited interpretability: they can detect which sets of populations have evidence for polygenic adaptation, but are unable to reveal where in the history of multiple populations these processes occurred. To address this, we created a method to detect polygenic adaptation in an admixture graph, which is a representation of the historical divergences and admixture events relating different populations through time. We developed a Markov chain Monte Carlo (MCMC) algorithm to infer branch-specific parameters reflecting the strength of selection in each branch of a graph. Additionally, we developed a set of summary statistics that are fast to compute and can indicate which branches are most likely to have experienced polygenic adaptation. We show via simulations that this method—which we call PolyGraph—has good power to detect polygenic adaptation, and applied it to human population genomic data from around the world. We also provide evidence that variants associated with several traits, including height, educational attainment, and self-reported unibrow, have been influenced by polygenic adaptation in different populations during human evolution.
- Novembre, J., & Barton, N. H. (2018). Tread lightly interpreting polygenic tests of selection. Genetics, 208(4), 1351.
Comment on the above.
- Sanjak, J. S., Sidorenko, J., Robinson, M. R., Thornton, K. R., & Visscher, P. M. (2018). Evidence of directional and stabilizing selection in contemporary humans. Proceedings of the National Academy of Sciences, 115(1), 151-156.
Modern molecular genetic datasets, primarily collected to study the biology of human health and disease, can be used to directly measure the action of natural selection and reveal important features of contemporary human evolution. Here we leverage the UK Biobank data to test for the presence of linear and nonlinear natural selection in a contemporary population of the United Kingdom. We obtain phenotypic and genetic evidence consistent with the action of linear/directional selection. Phenotypic evidence suggests that stabilizing selection, which acts to reduce variance in the population without necessarily modifying the population mean, is widespread and relatively weak in comparison with estimates from other species.
- Zeng, J., De Vlaming, R., Wu, Y., Robinson, M. R., Lloyd-Jones, L. R., Yengo, L., … & Powell, J. E. (2018). Signatures of negative selection in the genetic architecture of human complex traits. Nature genetics, 50(5), 746-753.
We develop a Bayesian mixed linear model that simultaneously estimates single-nucleotide polymorphism (SNP)-based heritability, polygenicity (proportion of SNPs with nonzero effects), and the relationship between SNP effect size and minor allele frequency for complex traits in conventionally unrelated individuals using genome-wide SNP data. We apply the method to 28 complex traits in the UK Biobank data (N = 126,752) and show that on average, 6% of SNPs have nonzero effects, which in total explain 22% of phenotypic variance. We detect significant (P < 0.05/28) signatures of natural selection in the genetic architecture of 23 traits, including reproductive, cardiovascular, and anthropometric traits, as well as educational attainment. The significant estimates of the relationship between effect size and minor allele frequency in complex traits are consistent with a model of negative (or purifying) selection, as confirmed by forward simulation. We conclude that negative selection acts pervasively on the genetic variants associated with human complex traits.
- Srinivasan, S., Bettella, F., Frei, O., Hill, W. D., Wang, Y., Witoelar, A., … & Deary, I. J. (2018). Enrichment of genetic markers of recent human evolution in educational and cognitive traits. Scientific reports, 8(1), 1-9.
Higher cognitive functions are regarded as one of the main distinctive traits of humans. Evidence for the cognitive evolution of human beings is mainly based on fossil records of an expanding cranium and an increasing complexity of material culture artefacts. However, the molecular genetic factors involved in the evolution are still relatively unexplored. Here, we investigated whether genomic regions that underwent positive selection in humans after divergence from Neanderthals are enriched for genetic association with phenotypes related to cognitive functions. We used genome wide association data from a study of college completion (N = 111,114), one of educational attainment (N = 293,623) and two different studies of general cognitive ability (N = 269,867 and 53,949). We found nominally significant polygenic enrichment of associations with college completion (p = 0.025), educational attainment (p = 0.043) and general cognitive ability (p = 0.015 and 0.025, respectively), suggesting that variants influencing these phenotypes are more prevalent in evolutionarily salient regions. The enrichment remained significant after controlling for other known genetic enrichment factors, and for affiliation to genes highly expressed in the brain. These findings support the notion that phenotypes related to higher order cognitive skills typical of humans have a recent genetic component that originated after the separation of the human and Neanderthal lineages.
p-hacked!
- Beissinger, T., Kruppa, J., Cavero, D., Ha, N. T., Erbe, M., & Simianer, H. (2018). A simple test identifies selection on complex traits. Genetics, 209(1), 321-333.
Important traits in agricultural, natural, and human populations are increasingly being shown to be under the control of many genes that individually contribute only a small proportion of genetic variation. However, the majority of modern tools in quantitative and population genetics, including genome-wide association studies and selection-mapping protocols, are designed to identify individual genes with large effects. We have developed an approach to identify traits that have been under selection and are controlled by large numbers of loci. In contrast to existing methods, our technique uses additive-effects estimates from all available markers, and relates these estimates to allele-frequency change over time. Using this information, we generate a composite statistic, denoted which can be used to test for significant evidence of selection on a trait. Our test requires pre- and postselection genotypic data but only a single time point with phenotypic information. Simulations demonstrate that is powerful for identifying selection, particularly in situations where the trait being tested is controlled by many genes, which is precisely the scenario where classical approaches for selection mapping are least powerful. We apply this test to breeding populations of maize and chickens, where we demonstrate the successful identification of selection on traits that are documented to have been under selection.
- Guo, J., Wu, Y., Zhu, Z., Zheng, Z., Trzaskowski, M., Zeng, J., … & Yang, J. (2018). Global genetic differentiation of complex traits shaped by natural selection in humans. Nature communications, 9(1), 1-9.
There are mean differences in complex traits among global human populations. We hypothesize that part of the phenotypic differentiation is due to natural selection. To address this hypothesis, we assess the differentiation in allele frequencies of trait-associated SNPs among African, Eastern Asian, and European populations for ten complex traits using data of large sample size (up to ~405,000). We show that SNPs associated with height (P=2.46×10−5), waist-to-hip ratio (P=2.77×10−4), and schizophrenia (P=3.96×10−5) are significantly more differentiated among populations than matched “control” SNPs, suggesting that these trait-associated SNPs have undergone natural selection. We further find that SNPs associated with height (P=2.01×10−6) and schizophrenia (P=5.16×10−18) show significantly higher variance in linkage disequilibrium (LD) scores across populations than control SNPs. Our results support the hypothesis that natural selection has shaped the genetic differentiation of complex traits, such as height and schizophrenia, among worldwide populations.
This one is curious in that it finds evidence of divergent selection in educational attainment SNPs, but then does not discuss it in the text!
- Uricchio, L. H., Kitano, H. C., Gusev, A., & Zaitlen, N. A. (2019). An evolutionary compass for detecting signals of polygenic selection and mutational bias. Evolution letters, 3(1), 69-79.
Selection and mutation shape the genetic variation underlying human traits, but the specific evolutionary mechanisms driving complex trait variation are largely unknown. We developed a statistical method that uses polarized genome‐wide association study (GWAS) summary statistics from a single population to detect signals of mutational bias and selection. We found evidence for nonneutral signals on variation underlying several traits (body mass index [BMI], schizophrenia, Crohn’s disease, educational attainment, and height). We then used simulations that incorporate simultaneous negative and positive selection to show that these signals are consistent with mutational bias and shifts in the fitness‐phenotype relationship, but not stabilizing selection or mutational bias alone. We additionally replicate two of our top three signals (BMI and educational attainment) in an external cohort, and show that population stratification may have confounded GWAS summary statistics for height in the GIANT cohort. Our results provide a flexible and powerful framework for evolutionary analysis of complex phenotypes in humans and other species, and offer insights into the evolutionary mechanisms driving variation in human polygenic traits.
- Sohail, M., Maier, R. M., Ganna, A., Bloemendal, A., Martin, A. R., Turchin, M. C., … & Neale, B. (2019). Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife, 8, e39702.
Genetic predictions of height differ among human populations and these differences have been interpreted as evidence of polygenic adaptation. These differences were first detected using SNPs genome-wide significantly associated with height, and shown to grow stronger when large numbers of sub-significant SNPs were included, leading to excitement about the prospect of analyzing large fractions of the genome to detect polygenic adaptation for multiple traits. Previous studies of height have been based on SNP effect size measurements in the GIANT Consortium meta-analysis. Here we repeat the analyses in the UK Biobank, a much more homogeneously designed study. We show that polygenic adaptation signals based on large numbers of SNPs below genome-wide significance are extremely sensitive to biases due to uncorrected population stratification. More generally, our results imply that typical constructions of polygenic scores are sensitive to population stratification and that population-level differences should be interpreted with caution.
- Berg, J. J., Harpak, A., Sinnott-Armstrong, N., Joergensen, A. M., Mostafavi, H., Field, Y., … & Coop, G. (2019). Reduced signal for polygenic adaptation of height in UK Biobank. Elife, 8, e39725.
Several recent papers have reported strong signals of selection on European polygenic height scores. These analyses used height effect estimates from the GIANT consortium and replication studies. Here, we describe a new analysis based on the the UK Biobank (UKB), a large, independent dataset. We find that the signals of selection using UKB effect estimates are strongly attenuated or absent. We also provide evidence that previous analyses were confounded by population stratification. Therefore, the conclusion of strong polygenic adaptation now lacks support. Moreover, these discrepancies highlight (1) that methods for correcting for population stratification in GWAS may not always be sufficient for polygenic trait analyses, and (2) that claims of differences in polygenic scores between populations should be treated with caution until these issues are better understood.
Together with the Sohail et al paper above, this is an important critical contribution about methods for dealing with “population stratification”.
- Uricchio, L. H. (2020). Evolutionary perspectives on polygenic selection, missing heritability, and GWAS. Human genetics, 139(1), 5-21.
Genome-wide association studies (GWAS) have successfully identified many trait-associated variants, but there is still much we do not know about the genetic basis of complex traits. Here, we review recent theoretical and empirical literature regarding selection on complex traits to argue that “missing heritability” is as much an evolutionary problem as it is a statistical problem. We discuss empirical findings that suggest a role for selection in shaping the effect sizes and allele frequencies of causal variation underlying complex traits, and the limitations of these studies. We then use simulations of selection, realistic genome structure, and complex human demography to illustrate the results of recent theoretical work on polygenic selection, and show that statistical inference of causal loci is sharply affected by evolutionary processes. In particular, when selection acts on causal alleles, it hampers the ability to detect causal loci and constrains the transferability of GWAS results across populations. Last, we discuss the implications of these findings for future association studies, and suggest that future statistical methods to infer causal loci for genetic traits will benefit from explicit modeling of the joint distribution of effect sizes and allele frequencies under plausible evolutionary models.
- Refoyo-Martínez, A., Liu, S., Jørgensen, A. M., Jin, X., Albrechtsen, A., Martin, A. R., & Racimo, F. (2020). How robust are cross-population signatures of polygenic adaptation in humans?. BioRxiv.
Over the past decade, summary statistics from genome-wide association studies (GWAS) have been used to detect and quantify polygenic adaptation in humans. Several studies have reported signatures of natural selection at sets of SNPs associated with complex traits, like height and body mass index. However, more recent studies suggest that some of these signals may be caused by biases from uncorrected population stratification in the GWAS data with which these tests are performed. Moreover, past studies have predominantly relied on SNP effect size estimates obtained from GWAS panels of European ancestries, which are known to be poor predictors of phenotypes in non-European populations. Here, we collated GWAS data from multiple anthropometric and metabolic traits that have been measured in more than one cohort around the world, including the UK Biobank, FINRISK, Chinese NIPT, Biobank Japan, APCDR and PAGE. We then evaluated how robust signals of polygenic adaptation are to the choice of GWAS cohort used to identify associated variants and their effect size estimates, while using the same panel to obtain population allele frequencies (The 1000 Genomes Project). We observe many discrepancies across tests performed on the same phenotype and find that association studies performed using multiple different cohorts, like meta-analyses, tend to produce scores with strong overdispersion across populations. This results in apparent signatures of polygenic adaptation which are not observed when using effect size estimates from biobank-based GWAS of homogeneous ancestries. Indeed, we were able to artificially create score overdispersion when taking the UK Biobank cohort and simulating a meta-analysis on multiple subsets of the cohort. This suggests that extreme caution should be taken in the execution and interpretation of future tests of polygenic adaptation based on population differentiation, especially when using summary statistics from GWAS meta-analyses.
- Stern, A. J., Speidel, L., Zaitlen, N. A., & Nielsen, R. (2020). Disentangling selection on genetically correlated polygenic traits using whole-genome genealogies. bioRxiv.
We present a full-likelihood method to estimate and quantify polygenic adaptation from contemporary DNA sequence data. The method combines population genetic DNA sequence data and GWAS summary statistics from up to thousands of nucleotide sites in a joint likelihood function to estimate the strength of transient directional selection acting on a polygenic trait. Through population genetic simulations of polygenic trait architectures and GWAS, we show that the method substantially improves power over current methods. We examine the robustness of the method under uncorrected GWAS stratification, uncertainty and ascertainment bias in the GWAS estimates of SNP effects, uncertainty in the identification of causal SNPs, allelic heterogeneity, negative selection, and low GWAS sample size. The method can quantify selection acting on correlated traits, fully controlling for pleiotropy even among traits with strong genetic correlation (|rg| = 80%; c.f. schizophrenia and bipolar disorder) while retaining high power to attribute selection to the causal trait. We apply the method to study 56 human polygenic traits for signs of recent adaptation. We find signals of directional selection on pigmentation (tanning, sunburn, hair, P=5.5e-15, 1.1e-11, 2.2e-6, respectively), life history traits (age at first birth, EduYears, P=2.5e-4, 2.6e-4, respectively), glycated hemoglobin (HbA1c, P=1.2e-3), bone mineral density (P=1.1e-3), and neuroticism (P=5.5e-3). We also conduct joint testing of 137 pairs of genetically correlated traits. We find evidence of widespread correlated response acting on these traits (2.6-fold enrichment over the null expectation, P=1.5e-7). We find that for several traits previously reported as adaptive, such as educational attainment and hair color, a significant proportion of the signal of selection on these traits can be attributed to correlated response, vs direct selection (P=2.9e-6, 1.7e-4, respectively). Lastly, our joint test uncovers antagonistic selection that has acted to increase type 2 diabetes (T2D) risk and decrease HbA1c (P=1.5e-5).
- Lawson, D. J., Davies, N. M., Haworth, S., Ashraf, B., Howe, L., Crawford, A., … & Timpson, N. J. (2020). Is population structure in the genetic biobank era irrelevant, a challenge, or an opportunity?. Human Genetics, 139(1), 23-41.
Replicable genetic association signals have consistently been found through genome-wide association studies in recent years. The recent dramatic expansion of study sizes improves power of estimation of effect sizes, genomic prediction, causal inference, and polygenic selection, but it simultaneously increases susceptibility of these methods to bias due to subtle population structure. Standard methods using genetic principal components to correct for structure might not always be appropriate and we use a simulation study to illustrate when correction might be ineffective for avoiding biases. New methods such as trans-ethnic modeling and chromosome painting allow for a richer understanding of the relationship between traits and population structure. We illustrate the arguments using real examples (stroke and educational attainment) and provide a more nuanced understanding of population structure, which is set to be revisited as a critical aspect of future analyses in genetic epidemiology. We also make simple recommendations for how problems can be avoided in the future. Our results have particular importance for the implementation of GWAS meta-analysis, for prediction of traits, and for causal inference.
-
Chen, M., & Chiang, C. W. (2020). Allele frequency differentiation at height-associated SNPs among continental human populations. bioRxiv.
Polygenic adaptation is thought to be an important mechanism of phenotypic evolution in humans, although recent evidence of confounding due to residual stratification in consortium GWAS made studies of polygenic adaptation more difficult to interpret. Using FST as a measure of allele frequency differentiation, a previous study has shown that the mean FST among African, East Asian, and European populations is significantly higher at height-associated SNPs than that found at matched non-associated SNPs, suggesting that polygenic adaptation is one of the reasons for differences in human height among these continental populations. However, we showed here even though the height-associated SNPs were identified using only European ancestry individuals, the estimated effect sizes are significantly associated with structures across continental populations, potentially explaining the elevated level of differentiation previously reported. To alleviate concerns of biased ascertainment of SNPs, we re-examined the distribution of FST at height-associated alleles ascertained from two biobank level GWAS (UK Biobank, UKB, and Biobank Japan, BBJ). We showed that when compared to non-associated SNPs, height-associated SNPs remain significantly differentiated among African, East Asian, and European populations from both 1000 Genomes (p = 0.0012 and p = 0.0265 when height SNPs were ascertained from UKB and BBJ, respectively), and Human Genome Diversity Panels (p = 0.0225 for UKB and p = 0.0032 for BBJ analyses). In contrast to FST-based analyses, we found no significant difference or consistent ranked order among continental populations in polygenic height scores constructed from SNPs ascertained from UKB and BBJ. In summary, our results suggest that, consistent with previous reports, height-associated SNPs are significantly differentiated in frequencies among continental populations after removing concerns of confounding by uncorrected stratification. Polygenic score-based analysis in this context appears to be susceptible to the choice of SNPs and, as we compared to FST-based statistics in simulations, would lose power in detecting polygenic adaptation if there are independent converging selections in more than one population.
Notable follow-up to Guo et al studies.
I want to close with an honorable mention:
-
Wu, D. D., & Zhang, Y. P. (2011). Different level of population differentiation among human genes. BMC evolutionary biology, 11(1), 1-7.
Background
During the colonization of the world, after dispersal out of African, modern humans encountered changeable environments and substantial phenotypic variations that involve diverse behaviors, lifestyles and cultures, were generated among the different modern human populations.
Results
Here, we study the level of population differentiation among different populations of human genes. Intriguingly, genes involved in osteoblast development were identified as being enriched with higher FST SNPs, a result consistent with the proposed role of the skeletal system in accounting for variation among human populations. Genes involved in the development of hair follicles, where hair is produced, were also found to have higher levels of population differentiation, consistent with hair morphology being a distinctive trait among human populations. Other genes that showed higher levels of population differentiation include those involved in pigmentation, spermatid, nervous system and organ development, and some metabolic pathways, but few involved with the immune system. Disease-related genes demonstrate excessive SNPs with lower levels of population differentiation, probably due to purifying selection. Surprisingly, we find that Mendelian-disease genes appear to have a significant excessive of SNPs with high levels of population differentiation, possibly because the incidence and susceptibility of these diseases show differences among populations. As expected, microRNA regulated genes show lower levels of population differentiation due to purifying selection.
Conclusion
Our analysis demonstrates different level of population differentiation among human populations for different gene groups.
This 2011 study has not been replicated as far as I know, but it is highly interesting because it provides a general prior on what kind of genetic differences in traits to expect. Their central result:
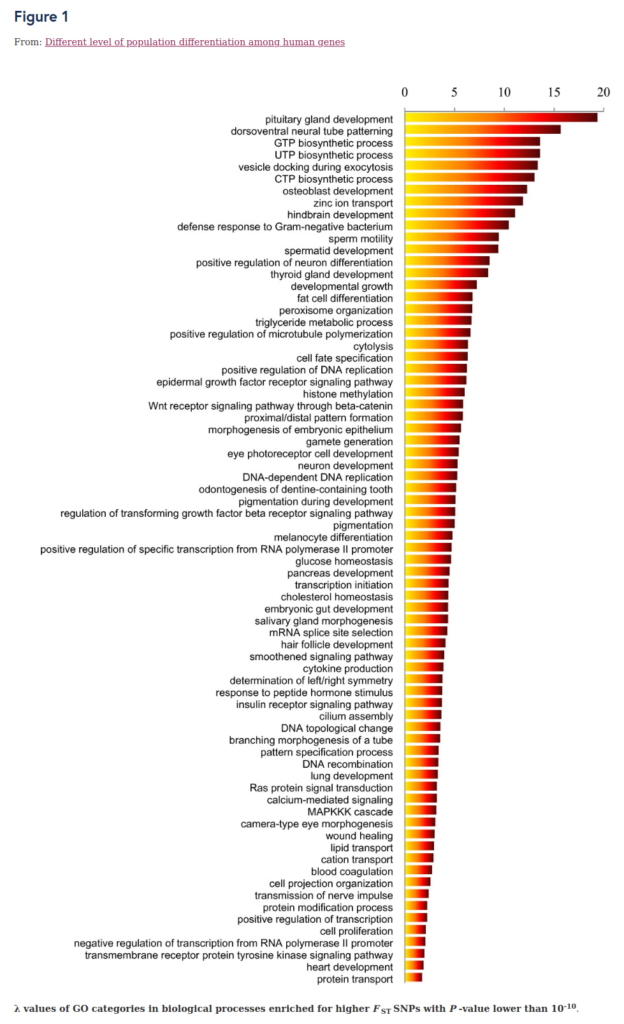
Skimming the list, one can see there are a lot of neurologically relevant pathways/processes enriched for divergent selection. This is expected on the general ground that human behavior results from the brain and adaptations to different climates and their resulting social systems requires behavioral changes. It would therefore be very interesting to see this study replicated with modern, larger datasets. This a general remark. Many of the studies above were done using the Okbay et al GWAS for education/intelligence, but there is now the Lee et al 2018 one, much larger. Thus, there is more power now to find selection signal, and also the possibility of using the sibling GWAS results from Lee et al (not part of their official release, but they are here).