If you follow population genetics on Twitter, you may have seen the current thing to be concerned about. For instance, we have Jonathan Pritchard at Stanford:
And Graham Coop at UC Davis:
We can quote Coop in full since he is most explicit:
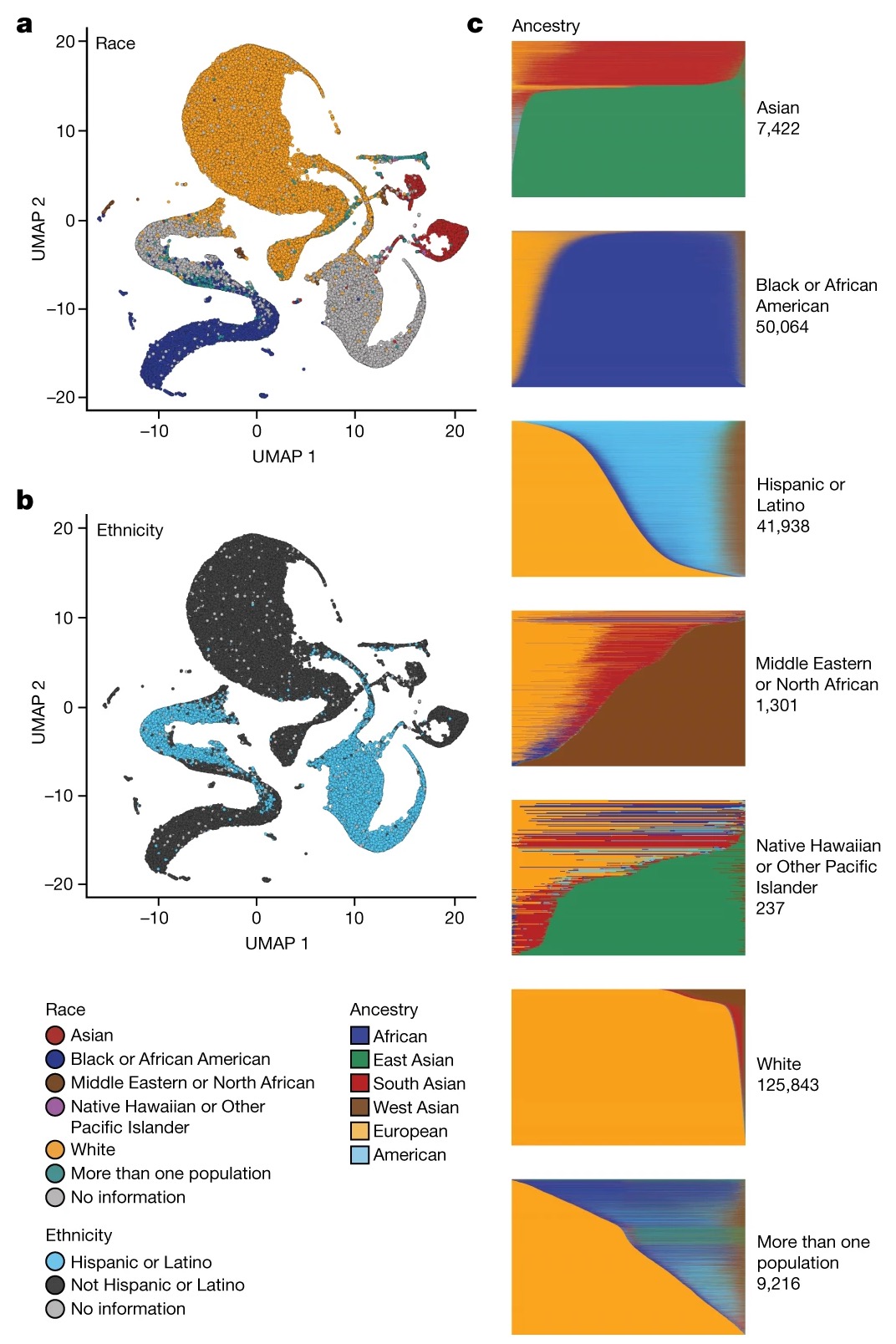
The All of Us paper is rightly being criticized for its UMAP figure, which suggests an overly discrete view of human variation—a problem that is compounded by colouring the plot with self-identified race and then omitting the “self-identified” from the title & legend. 1/n
The paper presents a major NIH resource, but it does not take on board the carefully thought-through advice of the NAS panel that the NIH commissioned (presumably for exactly this kind of purpose). 2/n
The admixture plots go some way to showing that people & self-id race do not map discretely onto clusters. But here again the choice to equate ancestry & self-identified race is misleading (e.g. choice of clustering level & colour matching in legend) 3/n
The authors state that these ancestry fractions match “six distinct and coherent ancestry groups” and that this “correspond[s] to participant self-identified race and ethnicity groups”. 4/n
There’s a whole set of additional issues raised here. Take for example the “American genetic ancestry.” which the authors say is “Latino/admixed american”… 5/n
Yet Latino/Hispanic are an ethnicities that usually includes people with a mixture of recent ancestors from Europe, Africa, and Indigenous American groups, and is not a distinct or separate grouping. 6/n
The “West Asia” genetic ancestry seems to be identified as “Middle Eastern” in the methods (again highlighting slippage). This ancestry looks to be present in a range of sample groups; is this a statistical artifact or.. 7/n
or is the inferred cluster absorbing North African and other genetic ancestries in some people? What are the downstream consequences of this? Are for example African-American participants going to be told they have “Middle Eastern ancestry”? 8/n
The authors elsewhere in the paper also continue to promote the use of discrete ancestry assignments to individuals, again risking conflating the race/ethnicity & genetic ancestry [see here for prev. critique https://twitter.com/Graham_Coop/status/1620105328225558528] 9/n
But their claims of concordance between ancestry & ethnicity only reflects the authors choosing to force a model of discrete categories on their data (see criticism below from a year ago 10/n https://twitter.com/Graham_Coop/status/1620105337213968385).
The authors’ decisions & analyses will confuse & mislead many, but the issues here run deep and will have many downstream consequences. 11/n
“All of Us” is a huge NIH resource, and there are many medical scientists who will rely on these results and decisions for downstream analyses/interpretation, meaning that errors made now will echo for years. That is far more damaging than just one UMAP plot. 12/n
So we have a bunch of worries, but the main point is that the authors were too explicit in linking self-identified race with genetics. As I quote tweeted:
Hilarious when one group of academics was slightly too honest about the connection between genetic ancestry and race with their plots. Much whining ensures to keep the party line defended.
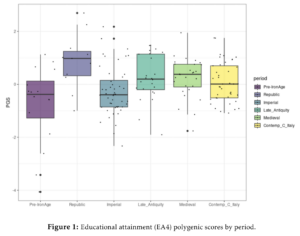
It is no surprise that these two variables correspond closely, since self-identified race is just the individual’s perception of their own race. Race is of course the same thing as genetic ancestry. How accurate is this? Consider these results from another large American study, the Add Health dataset:

This is a crude analysis where they computed each person’s largest ancestry fraction against their self-identified race to check for validity. Of the 5754 people who said they were non-Hispanic White (European Americans), 5644 also had most of their ancestry belong to the European cluster, that is, 98%. Of the 1940 who said they were non-Hispanic Black (African Americans), 1939 had their largest ancestry fraction as African, only 1 person had something else. A 99.9% match.
These results match what others have been finding for years, so they are not very surprising. I previously published a study using more refined methods to look at continuous variation in ancestry showing that this leads to excellent models which show that self-identified race and genetic race (ancestry) is just about the same thing with some self-report errors mixed in. 23andme did the same thing in their ancestry paper.

A model using only ancestry explained 94.13% of the variance in self-identified ancestry. When they authors added various other variables (age, sex) and their interactions with ancestry, the more complex models explained 94.16% of the variance. Clearly, these fine details were a waste of time, even if they have p < .05 in their dataset of 160k+ people.
If the topic was an ordinary one, no one would would be concerned about UMAP, or unhappy when a genetic model only explains 94% of the variance. But since the current mainstream defense against hereditarianism involves misleading people about the obvious link between race and genetics, they cannot do anything else in their position than complain about models with accuracy that would make any medical researcher jealous.
To be technical, we could say that they are right in a way. UMAP does a magic job of displaying many-dimensional data in only 2 dimensions plus colors, something that we puny humans can understand. It does this in ways that distort the data from the perspective of additive genetics. Nevertheless, we don’t care about this in any other field, so why care about it in genomics? It stinks of Isolated Demands for Rigor.
The new study is otherwise quite exciting:
Comprehensively mapping the genetic basis of human disease across diverse individuals is a long-standing goal for the field of human genetics1,2,3,4. The All of Us Research Program is a longitudinal cohort study aiming to enrol a diverse group of at least one million individuals across the USA to accelerate biomedical research and improve human health5,6. Here we describe the programme’s genomics data release of 245,388 clinical-grade genome sequences. This resource is unique in its diversity as 77% of participants are from communities that are historically under-represented in biomedical research and 46% are individuals from under-represented racial and ethnic minorities. All of Us identified more than 1 billion genetic variants, including more than 275 million previously unreported genetic variants, more than 3.9 million of which had coding consequences. Leveraging linkage between genomic data and the longitudinal electronic health record, we evaluated 3,724 genetic variants associated with 117 diseases and found high replication rates across both participants of European ancestry and participants of African ancestry. Summary-level data are publicly available, and individual-level data can be accessed by researchers through the All of Us Researcher Workbench using a unique data passport model with a median time from initial researcher registration to data access of 29 hours. We anticipate that this diverse dataset will advance the promise of genomic medicine for all.
It’s a big new dataset of 250k+ people with whole genome sequencing (WGS) data, and extensive phenotyping, including health records. The ancestry of the sample is very diverse, on purpose, so it is ideal for doing analyses of race differences in just about any phenotype of interest:
Genetic ancestry inference confirmed that 51.1% of the All of Us WGS dataset is derived from individuals of non-European ancestry. Briefly, the ancestry categories are based on the same labels used in gnomAD18. We trained a classifier on a 16-dimensional principal component analysis (PCA) space of a diverse reference based on 3,202 samples and 151,159 autosomal single-nucleotide polymorphisms. We projected the All of Us samples into the PCA space of the training data, based on the same single-nucleotide polymorphisms from the WGS data, and generated categorical ancestry predictions from the trained classifier (Methods). Continuous genetic ancestry fractions for All of Us samples were inferred using the same PCA data, and participants’ patterns of ancestry and admixture were compared to their self-identified race and ethnicity (Fig. 2 and Methods). Continuous ancestry inference carried out using genome-wide genotypes yields highly concordant estimates.
In fact, one might be tempted to congratulate researchers in the All of Us consortium with being quite free thinking with their questions in surveys:

Someone already made an appropriate meme to compare with the very boring variables in the UK Biobank:

Finally, it might be very difficult to use for anything too exciting because:
All of Us genomic data are available in a secure, access-controlled cloud-based analysis environment: the All of Us Researcher Workbench. Unlike traditional data access models that require per-project approval, access in the Researcher Workbench is governed by a data passport model based on a researcher’s authenticated identity, institutional affiliation, and completion of self-service training and compliance attestation28. After gaining access, a researcher may create a new workspace at any time to conduct a study, provided that they comply with all Data Use Policies and self-declare their research purpose. This information is regularly audited and made accessible publicly on the All of Us Research Projects Directory. This streamlined access model is guided by the principles that: participants are research partners and maintaining their privacy and data security is paramount; their data should be made as accessible as possible for authorized researchers; and we should continually seek to remove unnecessary barriers to accessing and using All of Us data.
Which is to say that you cannot download the data even if you go through all the academic hoops to get access, but must analyze it in their cloud system. For most researchers this is probably a benefit because then they don’t have to worry about installing genomics software, or having a big server for running the analyses. On the other hand, it also means it will be severely limited as some software won’t be available in their cloud computer, and there isn’t any easy way to run any unapproved analyses looking into the forbidden topics. This is a trend in genomics datasets in the past few years, where the censorship is built into the access to the datasets. Probably datasets used for more exciting work will have to be found in the private sector. Indeed, many people have been suggesting that a rogue billionaire or some consortium simply buy 23andme, now that its stock has crashed more than 90%, and release the data for all researchers to use:
While that seems a bit unlikely to happen right now (Metaculus?), in the coming years, with the price of sequencing data falling, there will eventually be huge, actually public access datasets with proper phenotyping that anyone can download and use for any purpose. The few existing public genetic datasets have seen massive usage rates since random graduate students don’t have to waste months trying to get access, maybe in vain. You simply click “Download” and start the sciencing. That’s how science is best served.