Woodley reminded me of the dysgenics for health outcomes by linking me to a study about the increasing rates of cancer. I had first reached this conclusion back in 2005 when I realized what it means for evolution that we essentially keep almost everyone alive despite their genetic defects. The problem is quite simple: mutations accumulate and mutations have net negative impact on the functioning of the body. Most of the genome appears not to be relevant for anything (‘junk’ / non-coding), so mutations in these areas don’t do anything. Of mutations that hit other areas, many of them are synonymous, and thus usually have no effect. Mutations in areas that matter generally have negative effects. Why? The human body is an intricate machine and it’s easier to fuck it up when you make random changes to the blueprint/recipe than improving upon it. So, basically, one has to get rid of the harmful mutations that happen and this is done via death and mate choice (preference for healthy partners), collectively: purifying selection. Humans still have mate choice and some natural selection, but the natural selection has been starkly reduced in strength since before medicine that actually works (i.e. not bloodletting etc.), and thus, by mutation-selection balance, the rates of genetic disorders and genetic dispositions for disease should increase. In other words, mutation load for disease in general should increase. Does it?
It’s not quite so simple to answer because of various confounders. The most important ones are improved diagnosing (we have better equipment to spot disorders now) and population aging (older people are more sick). Population aging can be avoided by compared same-aged samples measured in different times. Diagnosis changes are much harder to deal with, and one has to look for data where the diagnostic criteria either did not change for the period in question or changed in a way we can adjust for.
There’s also another issue. For a number of decades, we have been using a clever form of selection: prenatal screening (and preconception screening in some groups), which obviously selects against mutational load for the screened diseases. However, most of this testing is for aneuplodies (mostly Down’s) which usually results in sterile offspring and is thus irrelevant for mutational load for disease (because it is not contributed to the gene pool). However, some of the testing is for specific diseases, usually ones that happen to be quite prevalent in some racial group: Tay-Sachs etc. in Ashkenazis, Charlevoix-Saguenay etc. in Quebecians, Aspartylglucosaminuria etc. in Finns etc. One obviously cannot look for evidence of dysgenics for these diseases as the selection against them distorts the picture.
The studies
I didn’t do a thorough search. In fact, these were the first two studies I found plus the one Michael found. The point of this review is to bring the idea to your mind, not prove it conclusively with an exhaustive review.
Cancer
Cancer incidence increasing globally: The role of relaxed natural selection
Cancer incidence increase has multiple aetiologies. Mutant alleles accumulation in populations may be one of them due to strong heritability of many cancers. The opportunity for the operation of natural selection has decreased in the past ~150 years because of reduction of mortality and fertility. Mutation-selection balance may have been disturbed in this process and genes providing background for some cancers may have been accumulating in human gene pools. Worldwide, based on the WHO statistics for 173 countries the index of the opportunity for selection is strongly inversely correlated with cancer incidence in peoples aged 0-49 and in people of all ages. This relationship remains significant when GDP, life expectancy of older people (e50), obesity, physical inactivity, smoking and urbanization are kept statistically constant for fifteen (15) out of twenty-seven (27) individual cancers incidence rates. Twelve (12) cancers which are not correlated to relaxed natural selection after considering the six potential confounders are largely attributable to external causes like viruses and toxins. Ratios of the average cancer incidence rates of the 10 countries with highest opportunities for selection to the average cancer incidence rates of the 10 countries with lowest opportunities for selection are 2.3 (all cancers at all ages), 2.4 (all cancers in 0-49 years age group), 5.7 (average ratios of strongly genetically based cancers) and 2.1 (average ratios of cancers with less genetic background).
Coeliac disease
Increasing prevalence of coeliac disease over time
Background The number of coeliac disease diagnoses has increased in the recent past and according to screening studies, the total prevalence of the disorder is around 1%.
Aim To establish whether the increased number of coeliac disease cases reflects a true rise in disease frequency.
Methods The total prevalence of coeliac disease was determined in two population-based samples representing the Finnish adult population in 1978–80 and 2000–01 and comprising 8000 and 8028 individuals, respectively. Both clinically–diagnosed coeliac disease patients and previously unrecognized cases identified by serum endomysial antibodies were taken into account.
Results Only two (clinical prevalence of 0.03%) patients had been diagnosed on clinical grounds in 1978–80, in contrast to 32 (0.52%) in 2000–01. The prevalence of earlier unrecognized cases increased statistically significantly from 1.03% to 1.47% during the same period. This yields a total prevalence of coeliac disease of 1.05% in 1978–80 and 1.99% in 2000–01.
Conclusions The total prevalence of coeliac disease seems to have doubled in Finland during the last two decades, and the increase cannot be attributed to the better detection rate. The environmental factors responsible for the increasing prevalence of the disorder are issues for further studies.
Arthritis and other rheumatic conditions
Objective
To provide a single source for the best available estimates of the US prevalence of and number of individuals affected by osteoarthritis, polymyalgia rheumatica and giant cell arteritis, gout, fibromyalgia, and carpal tunnel syndrome, as well as the symptoms of neck and back pain. A companion article (part I) addresses additional conditions.
Methods
The National Arthritis Data Workgroup reviewed published analyses from available national surveys, such as the National Health and Nutrition Examination Survey and the National Health Interview Survey. Because data based on national population samples are unavailable for most specific rheumatic conditions, we derived estimates from published studies of smaller, defined populations. For specific conditions, the best available prevalence estimates were applied to the corresponding 2005 US population estimates from the Census Bureau, to estimate the number affected with each condition.
Results
We estimated that among US adults, nearly 27 million have clinical osteoarthritis (up from the estimate of 21 million for 1995), 711,000 have polymyalgia rheumatica, 228,000 have giant cell arteritis, up to 3.0 million have had self-reported gout in the past year (up from the estimate of 2.1 million for 1995), 5.0 million have fibromyalgia, 4–10 million have carpal tunnel syndrome, 59 million have had low back pain in the past 3 months, and 30.1 million have had neck pain in the past 3 months.
Conclusion
Estimates for many specific rheumatic conditions rely on a few, small studies of uncertain generalizability to the US population. This report provides the best available prevalence estimates for the US, but for most specific conditions more studies generalizable to the US or addressing understudied populations are needed.
Does it matter?
Yes. Treating diseases, especially rare diseases, is extremely expensive. As such, for countries with public health-care, there’s a very strong economic argument in favor of health eugenics via editing or embryo/gamete selection.
Socio-economic burden of rare diseases: A systematic review of cost of illness evidence
Cost-of-illness studies, the systematic quantification of the economic burden of diseases on the individual and on society, help illustrate direct budgetary consequences of diseases in the health system and indirect costs associated with patient or carer productivity losses. In the context of the BURQOL-RD project (“Social Economic Burden and Health-Related Quality of Life in patients with Rare Diseases in Europe”) we studied the evidence on direct and indirect costs for 10 rare diseases (Cystic Fibrosis [CF], Duchenne Muscular Dystrophy [DMD], Fragile X Syndrome [FXS], Haemophilia, Juvenile Idiopathic Arthritis [JIA], Mucopolysaccharidosis [MPS], Scleroderma [SCL], Prader-Willi Syndrome [PWS], Histiocytosis [HIS] and Epidermolysis Bullosa [EB]). A systematic literature review of cost of illness studies was conducted using a keyword strategy in combination with the names of the 10 selected rare diseases. Available disease prevalence in Europe was found to range between 1 and 2 per 100,000 population (PWS, a sub-type of Histiocytosis, and EB) up to 42 per 100,000 population (Scleroderma). Overall, cost evidence on rare diseases appears to be very scarce (a total of 77 studies were identified across all diseases), with CF (n=29) and Haemophilia (n=22) being relatively well studied, compared to the other conditions, where very limited cost of illness information was available. In terms of data availability, total lifetime cost figures were found only across four diseases, and total annual costs (including indirect costs) across five diseases. Overall, data availability was found to correlate with the existence of a pharmaceutical treatment and indirect costs tended to account for a significant proportion of total costs. Although methodological variations prevent any detailed comparison between conditions and based on the evidence available, most of the rare diseases examined are associated with significant economic burden, both direct and indirect.
Economic burden of common variable immunodeficiency: annual cost of disease
Objectives: In the context of the unknown economic burden imposed by primary immunodeficiency diseases, in this study, we sought to calculate the costs associated with the most prevalent symptomatic disease, common variable immunodeficiency (CVID). Methods: Direct, indirect and intangible costs were recorded for diagnosed CVID patients. Hidden Markov model was used to evaluate different disease-related factors and Monte Carlo method for estimation of uncertainty intervals. Results: The total estimated cost of diagnosed CVID is US$274,200/patient annually and early diagnosis of the disease can save US$6500. Hospital admission cost (US$25,000/patient) accounts for the most important expenditure parameter before diagnosis, but medication cost (US$40,600/patients) was the main factor after diagnosis primarily due to monthly administration of immunoglobulin. Conclusion: The greatest cost-determining factor in our study was the cost of treatment, spent mostly on immunoglobulin replacement therapy of the patients. It was also observed that CVID patients’ costs are reduced after diagnosis due to appropriate management.
There’s also lots of these kinds of studies, the second paper summarizes a number of them for this cluster of diseases:
A Spanish study reported that mean annual treatment costs for children and adult PID patients were e 6520 and 17,427, respectively. Total treatment costs spent on IVIg therapy proce- dures in Spain were approximately e 91.8 million annually, of which 94% consisted of drug cost [27] . Another study conducted in Belgium estimated the annual costs for IVIg therapy on an average to be e 12,550 [28].
Galli et al . [29] assessed the economic impact associated with method of treatment of PID patients in Italy. Regarding the monthly treatment costs associated with the treatment of a typ- ical 20 kg child, the study reported antibiotic therapy to cost of e 58,000, Ig cost of e 468,000 and patients ’ hospitalizations cost of e 300,000 for IVIg method.
Haddad et al . [26] conducted a cost analysis study in the French setting and reported the total monthly treatment cost for a patient using hospital-based 20 g IVIg to be e 1192.19, in which approximately 57% of the total treatment cost was spent on Ig preparation and 39% on hospital admission charges. Another investigation on French PID patients demonstrated the yearly cost of hospital-based IVIg to be e 26,880 per patient [30] .
Other cost analysis studies comparing the direct cost impacts of Ig replacement methods reported annual per patient costs for hospital-based IVIg were US$14,124 in Sweden [31] , e 31,027 and e 17,329 for adults and children in Germany, respectively [32] , and e 18,600 in UK [33] . On the basis of one Canadian study, we found that total annual base case expenditure for hospital-based IVIg therapy of children and adults were $14,721 and $23,037 (in Canadian dollars), respectively. The annual per patient cost of Ig was 75%, the cost of physician and nurse care and hospital admission was 16% and the cost of time lost because of treatment was 8% [34]
The Genomic Health Of Ancient Hominins?
Davide Piffer reminded me that there is study of ancient genomes’ health, which finds that:
The genomes of ancient humans, Neandertals, and Denisovans contain many alleles that influence disease risks. Using genotypes at 3180 disease-associated loci, we estimated the disease burden of 147 ancient genomes. After correcting for missing data, genetic risk scores were generated for nine disease categories and the set of all combined diseases. These genetic risk scores were used to examine the effects of different types of subsistence, geography, and sample age on the number of risk alleles in each ancient genome. On a broad scale, hereditary disease risks are similar for ancient hominins and modern-day humans, and the GRS percentiles of ancient individuals span the full range of what is observed in present day individuals. In addition, there is evidence that ancient pastoralists may have had healthier genomes than hunter-gatherers and agriculturalists. We also observed a temporal trend whereby genomes from the recent past are more likely to be healthier than genomes from the deep past. This calls into question the idea that modern lifestyles have caused genetic load to increase over time. Focusing on individual genomes, we find that the overall genomic health of the Altai Neandertal is worse than 97% of present day humans and that Otzi the Tyrolean Iceman had a genetic predisposition to gastrointestinal and cardiovascular diseases. As demonstrated by this work, ancient genomes afford us new opportunities to diagnose past human health, which has previously been limited by the quality and completeness of remains.
The authors themselves note the connection to the proposed recent dysgenic selection:
The genomic health of ancient individuals appears to have improved over time (Figure 3B). This calls into question the idea that genetic load has been increasing in human populations (Lynch 2016). However, there exists a perplexing pattern: ancient individuals who lived within the last few thousand years have healthier genomes, on average, than present day humans. This deviation from the observed temporal trend of improved genomic health opens up the possibility that deleterious mutations have accumulated in human genomes in the recent past. The data presented here do not provide adequate information to address this hypothesis, which we leave for future follow-up studies.
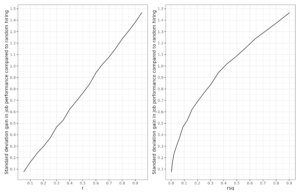
In other words, we expect the recent pattern to look something like this:



This is officially the first Disqus comment made for the site. Testing to see if it works. If all goes well, might enable comments for all the older posts (maybe skip a few to avoid annoying culture wars/SJW idiots).
If I end up spending too much time moderating comments, I will turn them off again.
Fred Wilson designated two readers to be moderators on his AVC.com blog, which uses Disqus as well (his firm is an investor in Disqus).